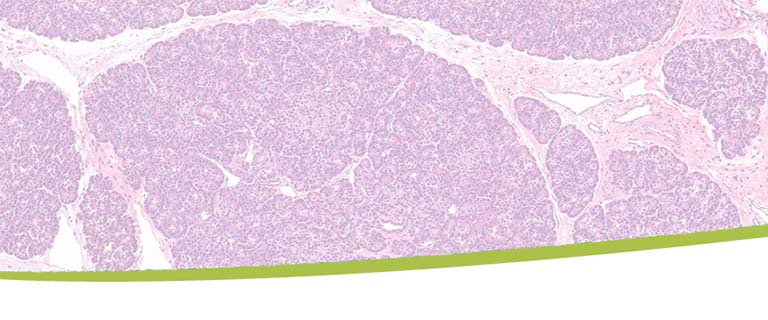
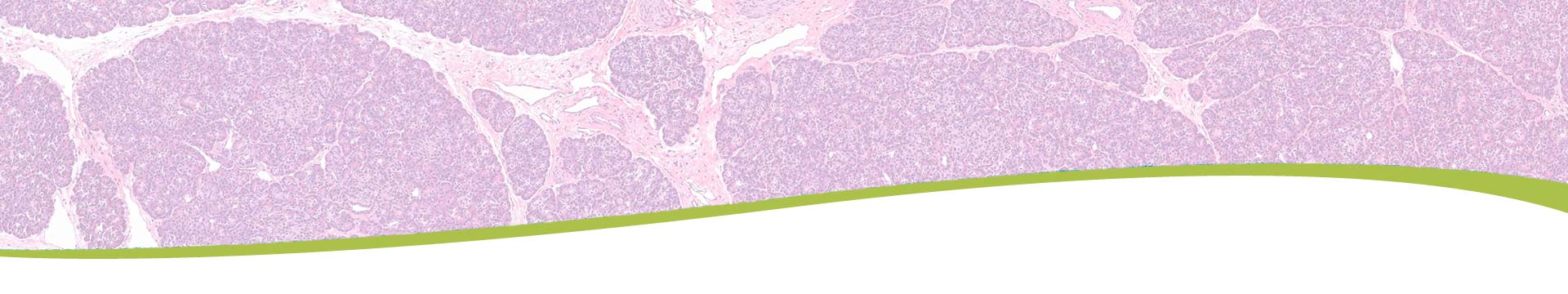
Alpha Cell Physiology & Dysfunction
-

Nicolai Doliba & Doris Stoffers
University of Pennsylvania
Role of Alpha Cells in Pathogenesis of Type 1 DiabetesMultiple islet autoantibodies (AAb+) predict type 1 diabetes (T1D) and hyperglycemia within 10 years. By contrast, T1D develops in just ~15% of single AAb+ (generally against glutamic acid decarboxylase, GADA+) individuals; hence the single AAb+ state may represent an early stage of T1D. We previously discovered the functional defects in suppression of glucagon secretion in non-diabetic, normoglycemic GADA+ donors which precedes the insulin secretory defect seen in T1D. This GADA+ donor alpha cell phenotype is accompanied by altered cAMP signaling and alpha cell gene expression, particularly in glycolysis and oxidative phosphorylation. The mechanism of this glucagon secretion defect in GADA+ alpha cells remains poorly understood. Therefore, the goal of the current project is to test whether the reduced or lost suppression of glucagon secretion by glucose in alpha cells during the progression of T1D is causally related to alteration of the cAMP pathway and/or bioenergetics.
This study will help to identify possible protective or pathogenic factors which may play a role in alpha cell dysfunction during development of T1D. These factors may then be used in the future for drug development to prevent or delay progression to T1D by targeting the early single AAb+ stage.
-

Vira Kravets and Edward James
University of California San Diego
Funded by: Diabetes Research Connection
Connection between alpha cell function and T-cell antigen specificity in type 1 diabetesGiven that in Aab+ donors alpha cells become dysfunctional before donor’s blood glucose levels are impaired, it is important to differentiate whether a) alpha cells become dysfunctional first, which attracts and activates immune cells, or b) vice versa. In pre – T1D the first phase insulin secretion is dampened. We and others have shown that this phase is triggered by “ first – responder ” beta cells – a stable beta cell subpopulation ( same cell or it ’ s neighbor remains 1 st responder for 24 – to – 48h with frequent restimulation ) . We also found that majority of the 1 st responders are alpha – cell – neighboring cells. Putting everything together, our question is whether alpha cells are targeted in T1D, which leads to the death /dysfunction of the neighboring beta cells ( 1 st responders ) first, and is manifested via loss of the first phase peak of GSIS in pre – T1D patients ? This question is not currently pursued by the mainstream research direction, but if answered, could change our view of T1D pathogenesis and suggest novel targets for therapy. We aim to study heterogeneity of alpha and beta cell function i n live pancreatic tissue slices. For this we will be using advanced NADH and Ca²⁺ imaging techniques . We hypothesize that, in early – stage T1D, some alpha – and their neighboring beta cells will show impaired metabolism and Ca²⁺ response to nutrients , compared to non – diabetic (ND) tissue . We will test how neoantigen – specific T – cell clones ( isolated & provided by James Lab) interact with alpha – and neighboring beta cells in live tissue slices in recent – onset T1D donors. We expect that , compared to rested (naive) clones, the pre – activated T – cell clones will behave differently : with altered mo tility profiles , proliferation, and frequency of contact with alpha cells and their neighboring beta c ells . Furthermore, w e wi ll manipulate alpha – T cell interactions using functional grade antibodies that block HLA presentation. For additional insights into the mechanism (add – value experiment) , we wi ll develop a staining protocol to use fluorescently labeled HLA tetramers complexed with known immunogenic peptides that are expressed in alpha cells (designed & provided by James Lab ) to tag T cells in T1D tissue . Using AI – assisted tools and 3D confocal microscopy, we wi ll analyze alp h a – T – cell immunologic synapse formation and networks to understand how T – cell specificity affects alpha – T cell interactions. -

Julia Panzer
City of Hope
Functional alpha cell heterogeneityIn type 1 diabetes (T1D), the destruction of insulin-producing beta cells is well known. But another emerging issue— not well understood—is the dysfunction of alpha cells. These cells produce glucagon, a hormone that helps prevent blood sugar from dropping too low. In people with T1D, alpha cells often fail to respond properly when blood sugar gets too low, which can lead to dangerous episodes of hypoglycemia. At the same time, they may release too much glucagon when it’s not needed, contributing to high blood sugar. Despite the critical role of alpha cells in managing blood sugar, we still know very little about how and why they go wrong in T1D.
Emerging research suggests that not all alpha cells are the same—there may be distinct groups with different roles. These unique behaviors may be linked to differences in how the cells are wired or which genes they express. The goal of this project is to understand these differences more clearly and find out how they change in T1D. To do this, I’ll use advanced imaging tools to study living human pancreatic tissue and isolated islets, allowing me to watch alpha cells in action. I’ll also analyze the molecular profiles of these cells to connect what they do with how they’re built.
By uncovering how alpha cell subtypes work—and how they fail in T1D—this research could open the door to better treatments. In the future, restoring proper alpha cell function could help prevent both low and high blood sugar in people with diabetes, improving safety and quality of life.
Beta Cell Physiology and Dysfunction
-

Andisheh Abedini & Ann Marie Schmidt
NYU School of Medicine
Uncovering pathological factors and mechanisms leading to β-cell death in type 1 diabetesWe hypothesize that β-cell toxicity in T1D can be induced via different pathways by distinct pathological ligands (AGEs, S100-proteins, amyloidogenic amylin) of the receptor for advanced glycation endproducts (RAGE) that become upregulated in the diabetic pancreas. We also hypothesize that C-peptide (+) T1D subjects may be more susceptible to amylin-induced β-cell toxicity. Amylin is co-produced /co-secreted with insulin by β-cells; subjects that retain the ability to produce/secrete insulin may also produce/secrete amylin. We will test out hypotheses in three successive stages: 1) Method validation, 2) Hypothesis-driven proof-of-concept testing, and 3) Mechanistic studies guided by findings in Stage-2. Having completed Stage-1 using nPOD control pancreas sections (T2D and nondiabetic), we are ready to test our hypothesis in 4 phenotypes of human subjects (C-peptide(+) T1D, C-peptide(-) T1D, T2D and non-diabetic controls) according to two synergistic specific aims. Aim-1: We will determine whether RAGE lignads (AGEs, S100-proteins, amyloidogenic amylin) become upregulated in T1D islets. Aim-2: We will determine whether islet amyloidosis correlates with C-peptide levels in T1D. IHC-IF will be used to quantify RAGE ligands/signaling molecules (amylin/amyloid, AGEs, RAGE, mDia1) and loss in β-cell/islet area and apoptosis (insulin/synaptophysin, cleaved capase3) in pancreas sections. RAGE ligands and signaling molecules (S100A12, RAGE, mDia1) will also be measured in frozen pancreas specimens by qRT-PCR, and in plasma (S100B) by ELISA. Upregulated ligands/mediators identified in Stage-2 will be the focus of mechanistic studies in Stage-3; which aim to identify mediators of oxidative stress (NOX1, NOX4, NOS), inflammation (CCL2, TNFα, ILβ, IL6, IL10) and apoptosis (bax) in frozen pancreas and spleen (control) specimens viz qRT-PCR. Results will be biostatically compared/contrasted/correlated. Successful completion of the proposed studies will identify key pathological factors and associated mechanisms by which RAGE contributes to β-cell/islet pathology in T1D, providing important insight into new drug targets for β-cell preservation and treatment/prevention of this disease.
-

Amin Ardestani and Mohammad Lotfollahi
Hull York Medical School
Funded by: DFG/Wellcome Trust
Investigating the role of Hippo pathway in Type 1 DiabetesThe prevalence of type 1 diabetes (T1D) is on the rise, posing a significant burden on healthcare systems. T1D results from the immune system mistakenly attacking insulin – producing pancreatic β – cells, necessitating lifelong insulin treatment and continuous monitoring of blood sugar levels. The complex inflammatory nature of T1D, coupled with substantial gaps in our understanding of the molecular drivers b ehind β – cell destruction, presents challenges in developing effective treatment strategies. The primary challenge this project tackles is the need for more effective approaches to treat T1D by addressing the underlying cause – the destruction of pancreatic β – cells. This requires a thorough understanding of the molecular mechanisms responsible for β – cell damage. This project centers around the role of the Hippo signalling pathway, known for its involvement in regulating organ size and tissue hemostasis. We were the first to demonstrate the pivotal role of the Hippo signaling pathway in β – cell apopt osis , regeneration, and function. Pilot data indicate that the terminal effector of the Hippo pathway, Yes – Associated Protein (YAP), typically repressed in mature β – cells, is upregulated in both exocrine and endocrine compartments of individuals with T1D, acti ng as a potential regulator of β – cell apoptosis and islet inflammation. Therefore, elevated expression of YAP is a novel metabolic aspect of T1D. We hypothesize that elucidating the molecular mechanisms linked to YAP – induced damage is crucial for the d evelopment of targeted therapies for T1D. The project’s primary objectives are to unravel the role of YAP and its molecular partners in T1D. These objectives include: (1) Identifying how YAP expression is activated in T1D and characterizing the cell types responsible for YAP upregulation. (2) Inve stigating the expression and cellular localization of YAP partners. (3) Determining spatial transcriptome profiling of YAP – expressing pancreatic cells in T1D to identify and characterize gene programs and signaling events shaping their niche identities. Utilizing the nPOD collection of healthy and diseased human pancreatic tissue, the project aims to unravel the molecular aspects of islet autoimmunity and β – cell destruction in T1D. This knowledge will serve as the foundation for new therapeutic strategies targeting the Hip po pathway to protect β – cells in T1D. -

Amin Ardestani & Mohammad Lotfollahi
University of Hull & Wellcome Sanger Institute
Generation of a Comprehensive Atlas of the Human Pancreas in Type 2 DiabetesType 2 diabetes (T2D) is one of the most common chronic conditions worldwide, affecting hundreds of millions of people. It develops when the pancreas cannot produce enough insulin and/ or when the body no longer responds to insulin effectively. While it is clear that insulin – producing β – cell s fail in T2D, the reasons for this failure remain poorly understood. Increasing evidence suggests that other parts of the pancreas, including the surrounding exocrine tissue and immune cells, may play a crucial role in t his process. Our project will generate the comprehensive “atlas” of the human pancreas in T2D. Using advanced single – nucleus and spatial technologies, we will study pancreatic tissues from people with and without T2D at unprecedented resolution. This approach will allow us to examine not only the islet cells , but also the exocrine, stromal, and immune compartments of the pancreas. By mapping how these cell types interact and change in T2D, we hope to uncover the networks and pathways that lead to β – cell dys function. To interpret these large datasets, we will employ cutting – edge artificial intelligence and machine learning methods. These tools will help us disentangle how the local pancreatic environment shapes disease and will enable us to predict which cellular inter actions might be driving β – cell failure. We will then confirm key findings directly in human tissue sections and in pancreatic slice models. In addition to T2D, we will study pancreatic samples from people with cystic fibrosis (CF). Many adults w ith CF develop a form of diabetes caused by changes in the pancreatic environment. Comparing T2D and CF will provide unique insight into how exocrine disease contributes to β – cell stress and failure, and will highlight shared or distinct mechanisms across conditions. Ultimately, this project will produce a valuable resource for the scientific community and help guide the development of therapies aimed at protecting and restoring insulin – producing cells. -

Peter Arvan & Leena Haataja
University of Michigan
Proinsulin biosynthesis and (mis)folding in type 1 diabetesA well – recognized feature of pancreatic ß – cells is their limited expression of endogenous antioxidants. From recent support by nPOD and HIRN/CBDS, my lab made the entirely novel observation, using metabolic labeling in pancreatic slices derived from 5 T1D donors, that disulfide – bond formation (i.e., folding) of newly – synthesized ( radio labeled) proinsulin is dramatically impaired with a surprisingly large fraction of fully – reduced or only partially oxidized proinsulin , which we found to be significantly different ( p < 0.0001) in pancreatic tissue f rom either nondiabetic or single autoantibody – positive individuals (this also differs from what we observed in our studies of a type 2 diabet e s model, namely, db/db mice — although we prefer, eventually, to check human T2D ). Our findings are completely unexpected, and run contrary to the hypothesis that the ER hyperoxidized in T1D ; instead the results suggest a state of reductive ER stress. We are able to phenocopy this (i.e., get the same) result by exposing isolated human islets to low doses of reducing agents that not only prevent efficient proinsulin disulfide bond formation but also activate a stress response known as ER stress/ISR . Initial peer review of our work liked the study but said : “ Sample size for T1D donors is inadequate for broad conclusions and the availability of nPOD samples appears uncertain . ” We hypothesize that a dys – coordination of “redox metabolism” results in an ER defect in T1D ß – cells ( in other inflammation – associated diseases , reductive ER stress has also been d escribed ). We seek addition nPOD slices from T1D donors in order to critically test this hypothesis using islets in situ . As a longer – term goal, we seek to remediate the proinsulin oxidation problem with pharmacologic agents that might restore redox balance within the ER . These aims offer a potential paradigm shift in our thinking about insulin secretory deficiency in the ß – cells of individuals suffering from T1D. -

Ronadip Banerjee
Johns Hopkins University School of Medicine
Funded by: NIH/NIDDK
Immediate-early gene transcription factor (IEG-TF) expression within human pancreatic islets during pregnancy and gestational diabetesPregnancy is a unique physiologic state in adult females that occurs and resolves over a defined time period. The acute metabolic stress it causes on the mother triggers adaptations with maternal pancreatic islets. Failure of normal islet adaptation is a major contributor to gestational diabetes (GDM).In rodents, insulin-producing beta-cells proliferate causing expansion of beta-cell mass. This expansion is defective in rodent models of GDM. Normally, the gestationally-expanded mass regresses in the early postpartum period. We and others have begun to identify gene expression changes that occur during pregnancy and postpartum in this model system, including how defects in specific genes or signaling pathways contribute to GDM. We recently identified a number genes within specific islet cell types whose expression changes in late pregnancy or postpartum. Amongst these were “immediate early genes” (IEGs) including several transcription factors (TFs) that may regulate survival (or cell death) of beta-cells in late pregnancy. In this proposed project, we will test the hypothesis that IEG-TFs identified in our rodent studies regulate beta-cell mass and function in human pregnancy, and may be dysregulated in GDM. To do so, we will perform immunohistochemical staining for specific IEG-TFs. We will co-stain with hormone markers (insulin, glucagon) to confirm cell type of expression, as well as with a marker of apoptosis (TUNEL) to see if IEG-TF expression correlates with apoptosis or survival of beta-cells in pregnancy or GDM.
-

Sharon Baumel-Alterzon
City of Hope
Funded by: NIH/NIDDK
Pancreatic Beta-Cells, Oxidative Stress, and Gestational DiabetesThe late stages of mammalian pregnancy are accompanied by a mild increase in insulin resistance, likely due to enhanced glucose demand for the growing fetus. Therefore, as an adaptive process to maintain normal blood glucose levels during pregnancy, maternal insulin – producing cell ( β – cell ) mass expands, leading to increased insulin release. Defects in functional β – cell adaptive expansion during pregnancy can lead to gestational diabetes mellitus (GDM). While the exact mechanisms that promote GDM are poorly understood, GDM is associated with inadequate functional β – cell mass expansion and with a systematic increase in oxidative stress (OS) . W e recently show ed that the levels of NRF2 , the master regulator of OS, are upregulated in mouse β – cells at gestational day 15 (Haidery et al. Redox Biology 2025) . Moreover, towards the end of pregnancy , mice with β – cell – specific Nrf2 delet ion (βNrf2KO) display reduced β – cell proliferation, increased β – cell OS and 4 – HNE ( 4 – Hydroxy – 2 – Nonenal , a lipid peroxidation product formed under OS) levels , compromised β – cell function, and elevated β – cell death, leading to impaired β – cell mass expansion and dysregulated glucose homeostasis. Importantly, the gestational hormone 17 – β – estradiol (E2) increases NRF2 levels, and downregulation of NRF2 suppresses E2 – induced protection of β – cells against OS , suggesting that E2 exerts its antioxidant effects through activation of NRF2 signaling in β – cells. Collectively, these data highlight the critical role of NRF2 in regulating OS during the adaptive response of β – cells in pregnancy and identify NRF2 as a potential therapeutic target for GDM treatment . W e propose further investigating NRF2 regulation in β – cells during human pregnancy. Specifically, we aim to determine whether, as observed in rodents, NRF2 levels and activity are upregulated in human β – cells during normal pregnancy and whether they are impaired in GDM . To test this, we plan to immuno label pancreatic tissues from pregnant (donors with normal pregnancy and GDM) and non – pregnant women using antibodies against (a) C – peptide and NRF2 ; (b) C – peptide and Sod1 (a Nrf2 target gene) ; (c) C – peptide and Nqo1 (another Nrf2 target gene); and (d) C – peptide and 4 – HNE. NRF2 levels and activity , as well as OS marker (4 – HNE) will be quantitated in β – cells. In summary , w e ai m to explor e how NRF2, a key factor that protects cells from oxidative stress, supports insulin – producing cells during pregnancy and whether its failure may play a role in gestational diabetes. -

Richard Benninger
University of Colorado Denver
Funded by: Breakthrough T1D
The role of protein kinase C delta in beta-cell death and dysfunction in type 1 diabetesThe goal of this study is identify novel mechanisms of immune-mediated β-cell death
and dysfunction such that these mechanisms can be exploited to protect against the
onset and progression of T1D. To achieve this goal, we propose the following aims: 1)
Determine if reduced electrical coupling, altered calcium signaling, and activation of
protein kinase C delta (PKCδ) occur in islets from newly diagnosed and autoantibody
positive donors compared to healthy donors 2) Determine if increased activation of
PKCδ correlates with β-cell apoptosis in islets from newly diagnosed and autoantibody
positive donors compared to healthy donors. The results of this study will identify a
novel role for decreased electrical coupling, altered calcium signaling, and activation of
PKCδ in mediating β-cell death and dysfunction in the pathogenesis of T1D. -

Susan Bonner-Weir & Cristina Aguayo-Mazzucato
Joslin Diabetes Center
Funded by: NIH
Aging Beta CellType 2 diabetes (T2D) increases with age with the majority of patients being above the fifth decade of life. These data underline the importance of studying how aging contributes to the reduced beta cell mass and dysfunctional insulin secretion found in T2D. However, the specific contribution of beta cell aging and senescence to diabetes has not been studied. Beta cells progress through different life stages, and, with age, the older subpopulation of cells becomes enriched. This enrichment has led us to identify specific markers of beta cell aging whose expression we have validated in mouse and human pancreas. We would like to further understand this process and its contribution to the development of T2D by exploring the expression of these markers in the pancreas of patients with T2D and lean and obese non-diabetic age-matched controls.
-

Lynne Coluccio & Hiroshi Tokuo
Boston University
Role of myosin-I in insulin secretionIn pancreatic beta cells, insulin is produced as proinsulin and packaged into secretory granules at a region of the cell called the trans-Golgi network (TGN). After maturation which includes the conversion of proinsulin to insulin, insulin granules are trafficked to the cell membrane where their contents are released through a process known as exocytosis. Actin filaments participate in different phases of insulin granule production and release. Our laboratory studies myosin motor proteins, which associate with actin filaments and modulate their activity. We have found that loss of the unconventional myosin Myo1b reduces insulin and proinsulin content. This results in less insulin secretion from Myo1b-depletedinsulinoma cells. Importantly, using an in situ fluorescent pulse-chase strategy to track nascent proinsulin, we found that loss of Myo1b impaired the early-stage trafficking of (pro)insulin granules. Our working model is that Myo1b is involved in early-stage insulin granule formation in beta cells. As diabetes is characterized by a reduction in insulin synthesis, insulin granule formation, and insulin secretion, our goal is to determine whether Myo1b loss is associated with the impaired insulin secretion in diabetic beta cells.
-

Yingfeng Deng
City of Hope
Uridine in the pathogenesis of T1DBlood uridine levels are elevated in T1D. However, its significance to the disease pathogenesis remains largely unexplored. Our pilot studies indicate uridine regulates glucose – stimulated insulin secretion (GSIS) in mouse and human islets. We will use huma n pancreas tissue sections and live pancreas slices to study how chronic elevation in blood uridine might affect beta cell function and survival in T1D. -

Yuval Dor & Benjamin Glaser
Hebrew University of Jerusalem
Markers of stress in diabetic isletsOur project seeks to understand the molecular basis for beta cell failure in human diabetes. We have discovered that excessive glucose metabolism in beta cells may lead to double strand breaks in the DNA and activation of the tumor suppressor gene p53. With the help of nPOD, we are studying the significance if this response in human type 2 diabetes patients. In our experiments we utilize archived material from human pancreata to examine the expression of multiple markers of the DNA damage response, to understand the nature of this response.
-
Yuval Dor & Elad Horwitz
Hebrew University, Jerusalem, Israel
Beta cell DNA double strand breaks in Type 1 diabetesEmerging evidence suggests beta cell dysfunction and failure precede autoimmune destruction during the development of type 1 diabetes, but the underlying mechanisms are not known. Defining and targeting the changes leading to impaired beta cell function may lead to clinical improvement and prolonged honeymoon period. We have recently discovered that in type 2 diabetes, beta cells suffer major damage to their DNA, and have partly delineated the pathway leading from high glucose levels to breaks in the DNA (Tornovsky, Cell Metabolism, 2014). While the consequences of DNA breaks in beta cells remain unknown, extensive information about the biology of DNA damage in other fields of biology strongly suggests that this is an important pathogenic process that may contribute to beta cell failure via multiple pathways. In unpublished preliminary experiments that form the basis for this proposal, we found increased DNA breaks in beta cells of human patients shortly after diagnosis of type 1 diabetes. We hypothesize that DNA damage precedes, and may contribute to, the development of autoimmune destruction of beta cells, and are seeking evidence for or against this hypothesis using nPOD material. Consistent with this hypothesis, previous studies have shown that DNA breaks can trigger an immune response, and that defects in the DNA repair machinery may lead to autoinflammatory disease.
We request funds to support experiments that will solidify the observation of DNA damage in beta cells shortly after diagnosis with type 1 diabetes, and will test whether DNA damage occurs in beta cells prior to clinical diagnosis. In addition, we will characterize the beta cells and islets that show DNA damage to understand what may control this phenomenon. To so, we will stain histological material of the pancreas from people at risk for type 1 diabetes (autoantibody positive donors) and shortly after diagnosis with type 1 diabetes (as well as controls) for markers of DNA damage, oxidative stress, replicative stress and immune attack. Together, these may point towards DNA damage as a new potential modulator of the development and progression of type 1 diabetes.
-

Decio Eizirik, Ihsane Marhfour & Anne Op de beeck
Universite Libre De Bruxelles, Belgium
Funded by: JDRFI
Characterization of beta cell ER stress associated to enteroviral infection in type 1 diabetesEnteroviral infections are associated with an increased risk to develop type 1 diabetes. Several stains of enteroviruses are able to infect beta cells leading to both inflammation and cell death, two major factors for the onset of type 1 diabetes (T1D). The cellular factors that modulate cellular permissiveness to enteroviral infection and beta cell death remain largely unknown. Since ER stress is involved in beta cell death and viral infections induce ER stress in other cell types, the aim of this project is to address the role of ER stress in virally induced beta cell death and local inflammation.
For this purpose, we will first determine whether components of the unfolded protein response (UPR) are induced during beta cell infection with a diabetogenic coxsackievirus (CVB5). Our preliminary data indicate an increase in CHOP, Bip and XBP1s mRNA expression in infected INS-1E cells. These results must be confirmed at the protein level and the activated UPR pathways further detailed.
Second, the role of the putative viral-induced ER stress in beta cell death and release of inflammatory mediators will be addressed by modulation of their expression through a silencing/overexpression approach and further evaluation of cell viability after CVB5 infection. These experiments, and the above described experiments, to be initially made in INS-1E cells, will be confirmed in primary rat and human beta cells. The accumulation of the involved ER stress markers in CVB5-infected beta cells, as well as co-detection of the capsid viral protein, will be evaluated by immunohistochemistry. To confirm the in vivo relevance of these observations, we will evaluate the co-localisation of the involved ER stress markers with viral capsid proteins in sections of pancreas from controls and coxsackievirus infected- and non infected-T1D donors. Altogether these results should shed light on the role of ER stress in beta cell death during coxsackievirus infection.
-

Feyza Engin
University of Wisconsin-Madison
Examination of ER stress markers in type 1 diabetes samples in humansThe endoplasmic reticulum (ER) is an organelle that is responsible for the proper folding of proteins and biosynthesis of lipids and steroids. Disruption of ER homeostasis leads to ER stress and activates a highly conserved adaptive network called the unfolded protein response (UPR). ER stress has been implicated in several diseases associated with protein folding defects and low grade chronic inflammation including neurodegenerative diseases, type2 diabetes, and atherosclerosis. The ability of UPR mediators to activate inflammatory and apoptotic signaling suggest ER stress might also be critical in development or progression of type1 diabetes. Despite increasing evidence that points to a critical role for ER stress in the pathogenesis of type1 diabetes, to date alterations in this pathway in human diabetic patients has not been demonstrated leading to a great scientific gap in understanding how organelle stress and related stress responses function in the pathogenesis of this disease. Our specific aim is to investigate the ER stress marker expression in the pancreata of human type1 diabetes and control samples by using immunofluorescence (IF) technique.
-

Carmella Evans-Molina, Tatsuyoshi Kono & Emily Sims
Indiana University School of Medicine
Funded by: NIH
Regulation of Beta Cell Secretory Pathway Calcium Homeostasis in Type 1 DiabetesCalcium plays a vital role in many processes that govern beta cell function, including the production, maturation, and regulated secretion of insulin. The fidelity of these processes depends on the maintenance of calcium subcompartments and their respective transmembrane gradients, which are organized at both the cellular and organelle level. Then endoplasmic reticulum, Golgi apparatus, and secretory granules comprise a functional module referred to as the beta cell secretory pathway, through which insulin mRNA is translated and preproinsulin protein is processed to mature insulin and packaged for exocytosis. In aggregate, these organelles also represent the largest intracellular depots of calcium. Calcium dyshomeostasis arising within the integrated secretory unit of the β cell is a characteristic of Type 1 diabetes and contributes to altered insulin production and processing and impaired β cell function and survival. The goal of this work is to define changes in the expression patterns of key molecular regulators of secretory pathway calcium and proinsulin processing.
-

Ji-Ming Feng
Louisiana State University
The cellular distribution of Golli protein in human pancreatic isletsThe objective of this application is to determine the cellular distribution of Golli protein in human pancreatic islets. Golli (gene expressed in oligodendrocyte lineage) is an alternatively spliced product of myelin basic protein (MBP) gene featured by the presence of a unique Golli domain which contains a 133-amino-acid sequence fused in the N-terminal of classic MBP. The Golli peptide share 79% homology between the human and the mouse. Our previous works in mouse have identified that Golli protein serves as an important regulator of Ca2+ release –activated Ca2+ (CRAC) channel in T lymphocytes and voltage-dependent calcium channel (VDCC) in oligodendrocytes and neurons. This application will start from our original findings in mice that: Golli protein is expressed in β cells of mouse Langerhans islet and plays a critical role in insulin secretion. We propose here to investigate cellular distribution of Golli protein in human pancreas (both normal and type-1 and -2 diabetic) tissues, aim to build up a direct correlation of our findings of Golli in rodents to the type-1 and type-2 diabetes in human.
-

Herbert Gaisano
University of Toronto
Funded by: Canadian Institutes of Health Research and Breakthrough T1D
Spatio-temporal visualization of immune and non-immune islet injury in Type 1 DiabetesType 1 Diabetes (T1D) generally results from a poorly understood autoimmune process that leads to selective destruction of pancreatic beta cells. Interestingly, beta cell function is often not correlated with mass and declines very early in the disease before symptom onset, but the mechanisms driving this are not known. Islet beta cells existing in a functional hierarchy, with a small proportion of key beta cells exerting a disproportional effect on overall islet insulin release. These key beta cells, termed ‘hub’ and ‘leader’ cells are the sources of calcium oscillations and coordinate connectivity between other ‘follower’ beta cells, thereby governing the amplitude and duration of insulin secretory granule release. Previous work has also shown that hub and leader beta cells are a stable beta cell subtypes that are sensitive to metabolic and inflammatory stress in vitro, but whether loss or dysfunction of these beta cell subtypes are causally linked to overall beta cell functional decline and inter-individual heterogeneity in T1D has not been explored. In this study, we propose to test the hypothesis that hub and leader beta cells are lost early on in T1D, which would lead to extensive heterogeneity in islet insulitis and functional beta cell mass both between individuals at the same disease stage and between different disease stages in the same individual. To test these ideas, we will carry outlive cell imaging and functional studies on live human donor pancreas slices from individuals at different disease stages (no diabetes/control, early-late autoimmunity, recent onset T1D) using genetically targeted probes to track calcium dynamics. We will apply computational approaches to characterize beta cell functional hierarchy in these slices, as well as use of optical probes to photo-label hub and leader populations in situ for analysis of their behavior in the autoimmune slice milieu. We propose to further characterize the inter-relationships between hub and leader beta cells, beta cell stress pathways and islet insulitis using fixed pancreas sections. Finally, using the live slice in situ model, we will explore how small molecules to that selectively beta cell stress pathways (including senescnece and unfolded protein response)impacts on islet functional hierarchy and connectivity. Together these studies will take us closer to understanding the contributions of beta cell heterogeneity to T1D heterogeneity.
-

Ivan Gerling
University of Tennessee Health Science Center
Funded by: Breakthrough T1D
Defining islet heterogeneity using single islet transcriptomicsThe goal of this project is to obtain transcriptome data from human islets using either hybridization arrays or RNA sequencing technology. We plan to obtain complete information about relative expression of all genes in islets from a number of different organ donor groups with an emphasis on: 1) donors with no autoantibodies or diabetes, 2) donors with autoantibodies but no diabetes, 3) donors with newly diagnosed type 1 diabetes, 4) donors with long-standing (over 5 years) type 1 diabetes, and, 5) donors with type 2 diabetes. The dataset will be analyzed to identify genes whose expression differs significantly between one or more of these donor groups. The goal is to define what pathways and processes uniquely define islets in each donor group, in order to gain new insights into the pathophysiology of diabetes and discover possible new drug targets. To further that goal each list of differentially expressed genes (distinguishing islets in one group from one or more of the other groups) will be subjected to a data mining process designed to define if the list is significantly enriched in genes that: 1) belong to specific pathways or gene ontologies; 2) are regulated by specific transcription factors; 3) are known to be influenced by specific drugs, proteins or other molecules. This will provide a global (systems level) definition of gene expression patterns in islets from each of these donor groups. However, we also intend to make this dataset available to other investigators who may want to examine the data for expression of genes relevant to their specific studies.
-

David Harlan
University of Massachusetts Medical School
Transcriptome analysis of highly purified islet cell populationsTherapies focused on sustaining or promoting endogenous insulin production for individuals at risk for or with T1D will require a thorough understanding of the gene expression patterns in islet α, β, and δ cells from control subjects and from individuals with T1D. Toward that end, we have developed methods to essentially purify α, β, and δ cells from human pancreata by intracellular staining for specified intracellular hormone content, sorting the populations by flow cytometry, then obtaining detailed transcriptomes from each population using next generation RNA sequencing. We propose to define the transcriptomes of isolated α, β, and δ cells from those with and without T1D and from β cells with varying HLA Class I expression (dim or bright) from subjects with T1D. We will define the changes in genes expressed by islet cells under the pathological conditions of T1D and specifically, from β and α cells hyper-expressing Class I proteins as a potential marker o a pro-inflammatory microenvironment with islets. These studies elucidate the β and α cell dysfunction in T1D and may provide targets for future therapeutic intervention(s) in T1D.
-
David Harlan & Sally Kent
University of Massachusetts Chan Medical School
Single Islet Functional Heterogeneity from control donors, auto antibody+ donors, and donors with recent onset type 1 diabetes (T1D)In T1D, pancreatic islets are infiltrated by lymphocytes (so called “insulitis”) and those T cells appear to drive β-cell dysfunction, and death. However, it has been long observed that not all islets within a T1D donor’s pancreas are infiltrated; even those in close proximity such that some islets have many infiltrating T cells while others appear quite pristine. We do not understand the reason for this heterogeneity. In addition, we do not understand the heterogeneity
of individual islet responses to glucose stimulation.In this proposal, we will complete the development of a micro-punch that will enable us to ‘punch out’ individual islets from donors of pancreatic slices and characterize their cellular constituents, transcriptional profiles, and function using an in vitro GSIS assay.
-

Pamela Itkin-Ansari, William Balch & Randal Kaufman
UCSD
Funded by: NIDDK HIRN
Insulin interacting proteins and stress markers in T1DWe have developed novel methods for proteomic study of beta-cells within native islets. Using
antibodies to proinsulin or insulin we reported the first Biosynthetic Interaction Network for insulin
(Pottekat, Cell Reports, 2013). Based upon our finding that many proteins which interact directly with
insulin are T1D GWAS candidates or proteins required for ER homeostasis, we propose to determine
how these proteins and others in their class are altered during progression to T1D. Moreover, based
upon recent evidence of ER stress in T1D islets and our identification of compounds which stabilize
the ER protein folding environment in vivo, we propose to test new strategies for preserving or
restoring beta-cell mass in T1D. -
Pamela Itkin-Ansari
Sanford-Burnham-Prebys Medical Discovery Institute
The role inflammatory signaling crosstalk on human exocrine and beta cellsThere is increasing evidence for cross-talk between the islet and exocrine compartments of the pancreas. Diabetes is a risk factor for exocrine atrophy, pancreatitis, and pancreatic cancer. Conversely, pancreatitis is a risk factor for diabetes. Until now our ability to study human endocrine and exocrine cells has been limited to isolated islets and isolated exocrine tissue. The tissue slice program provides a unique opportunity to simultaneously examine the immediate effects of exposure to inflammatory associated with diabetogenic or pancreatitis on both human beta cell and exocrine cell physiology. Here we will treat pancreas slices with the exocrine inflammatory molecule caerulein or the diabetogenic cytokines IL1beta and IFNgamma, measuring cell function and stress in both compartments. We previously showed that short term treatment of isolated exocrine cultures with caerulein induced BMP signaling, the transcriptional repressors ID1 and ID3 that promoted a potent proliferative response, as well as expression of inflammatory cytokines that may affect islets (Lee et. al. 2011a). In contrast to exocrine cells, human islets responded to ID3 with replication stress and DNA damage (Lee et. al 2011b). We also recently profiled gene expression in human islets exposed to a cytokine cocktail of IL1beta and IFNgamma, finding that the islets underwent ER stress and were induced to express chemokines that may well impact neighboring exocrine cells. In summary, the pancreas slice program enables the study of human endocrine/exocrine cross-talk in tissue for the first time.
-

Michael Kalwat & Travis Johnson
Indiana Biosciences Research Institute
Funded by: Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
Leveraging heterogeneity to identify markers of resilient β-cells in T1DWe a im to identify factors contributing to the destruction of β – cells in diabetes. In this project, we took advantage of publicly available bulk and single cell gene expression data sets from human islets of healthy and diabetic donors (T1D and T2D) . We use d deep transfer learning platform DEGAS 1 combined with this data to identify subsets of β – cells within human islet single cell RNA seq data whose transcriptomic signature correlates with that of diabetic islets . We determined the transcriptomic differences between high – and low – scoring β – cells within the human islet scRNAseq data. As part of this project, we identified the gene DLK1 as a candidate marker of β – cell resilience under diabetic stress. Our project will take two major approaches. 1) We will use antibodies to stain for the candidate protein DLK1 that we predict is expressed in β – cells that persist in the face of autoimmunity. 2 ) We will use spatial transcriptomics in non – diabetic, AAB+, and T1D human pan creas to measure the expression of candidate genes identified by DEGAS analysis and the spatial location of cells expressing these genes. By analyzing the gene expression patterns of these cells in we anticipate defining genes associated with different β – c ell states, spanning from resilient to vulnerable . We anticipate that this knowledge will help uncover new mechanisms to improve β – cell survival, escape autoimmunity, and explain why some β – cells persist but others do not. -

Heiko Lickert & Teresa Rodriguez-Calvo
Helmholtz Zentrum München
IGF receptor-like 1 and 2, new therapeutic targets for increasing insulin sensitivity and beta cell healthDiabetes mellitus is a complex and multifactorial disease characterized by progressive loss or dysfunction of the insulin-producing beta cells in the pancreas. This results in chronic hyperglycemia and systemic metabolic complications and, in the long-term, in multi-organ damages; together, these complications create enormous medical and social burdens as well as causing premature deaths. Today, over 422 million people worldwide have been diagnosed with diabetes and the number is expected to rise in the next 20 years. Currently no treatment can stop or revert diabetes progression, except bariatric surgery and islet transplantation. However, donor shortage and risks associated with life-long immunosuppression demand the development of alternative therapies. Increasing beta-cell health, proliferation and regeneration is disease modifying and has the potential to cure diabetes. In a screen for novel regulators of endocrinogenesis, we identified a previously uncharacterized regulator of insulin action, beta cell proliferation and blood glucose homeostasis. Experiments are ongoing to validate its usage as target to improve insulin sensitivity and to preserve β cell mass. Since the diabetic beta cells will be the target of the intervention, the nPOD samples will help us to discover changes in the receptor expression during the disease progression.
-

Amelia Linnemann
Indiana University
Regulation of Islet Autophagy in Type 1 DiabetesOxidative stress occurs when increased production of cellular oxidants (e.g. Reactive Oxygen Species, or ROS) is not compensated for by an increase in cellular antioxidants/antioxidant enzymes. A considerable body of evidence supports the conclusion that oxidative stress is a common feature of type 1 diabetes (T1D), and that the process of autophagy plays a key role in mitigation of ROS to reduce oxidative stress and promote -cell survival. Indeed, we recently found that coupling of autophagy to the antioxidant response could reduce-cell ROS, and protect against oxidative damage and apoptosis in an STZ model of diabetes in mice. Autophagy has long been known to play a critical role in -cell homeostasis and survival in the context of type 2 diabetes, however much less is known about the role that autophagy plays in the pathogenesis of type 1 diabetes, particularly in humans. Therefore, in this project we propose to analyze autophagy in islets from cadaveric donors with type 1 diabetes. Number, spatial organization of autophagosomes and lysosomes, and colocalization of these vesicles will be quantified. Further, since a specialized form of autophagy known as crinophagy is important for insulin homeostasis, we will also determine if lysosome changes are associated with the changes in proinsulin that have been recently reported in islets from type 1 diabetic individuals.
-

Amelia Linnemann
Indiana University School of Medicine
The role of beta cell lysosomal dysfunction in diabetes autoimmunityAs alterations in autophagic flux may contribute to T1D, we are seeking to determine if particular lysosomal components of beta cell autophagy participate in or dysregulate this essential process. The focus of this addendum is on the enzyme cathepsin H as specific genetic variants in this gene have been identified as T1D–associated SNPs. Through these studies, we will better define whether T1D-susceptible variants of genes encoding lysosomal enzymes modulate markers of autophagy in human pancreas tissue.
-
Amelia Linnemann & Carmella Evans-Molina
Indiana University School of Medicine
Funded by: NIH/NIDDK
Evaluation of the islet-immune interface in Cystic Fibrosis-related DiabetesCystic fibrosis (CF)-related diabetes (CFRD) is present in upwards of 20% of adolescents and 40-50% of adults with CF and is associated with significant glycemic variability, poor glycemic control, and unpredictable hypoglycemia. CFRD is increasing in prevalence as new disease-modifying therapies have led to a significant increase in life expectancy in those with CF. While the etiology of CFRD is poorly understood, pancreatic islet cells, responsible for the production of insulin, somatostatin, glucagon, and other hormones, are subject to immune-mediated inflammation in CFRD. In CF, chronic inflammation and infection may lead to tissue damage, fibrosis, and overall impaired pancreatic function, culminating in destruction of the insulin producing beta cells resulting in diabetes and impaired counterregulatory responses. Even in type 1 diabetes, an autoimmune disease with the similar feature of beta cell destruction, changes in the beta cell-immune cell interface during disease pathogenesis remain largely unknown. Therefore, understanding the role and composition of the pancreas landscape in the context of CF may provide clues to the pathogenesis of CFRD and our global understanding of how beta cells and immune cells interact. We recently used CO-Detection by indEXing (CODEX) imaging to elucidate islet cell composition and surrounding immune infiltrate to understand the role of CF in CFRD/ pancreatic function. Our preliminary analysis demonstrated a landscape of infiltration in the context of CFRD that could provide insight into pathogenesis. In this project, we propose to build on these results using the Xenium Prime 5K Human Pan Tissue & Pathways Panel to evaluate gene expression at the single cell level in fresh frozen pancreas tissue sections from the same organ donors used for CODEX. We expect this powerful side-by-side analysis to provide unprecedented knowledge regarding the beta cell-immune cell interface and its altered landscape in CFRD disease development.
-
Amelia Linnemann & Alberto Pugliese
Indiana University School of Medicine
Funded by: Breakthrough T1D
Oxidized Insulin as a Biomarker for Type 1 Diabetes PathogenesisReactive oxidants are generated during the metabolism of glucose and serve as important signaling messengers to trigger insulin secretion and beta cell expansion in response to elevated levels of glucose. However, chronic exposure to oxidants under hyperglycemic conditions causes chronic oxidative stress which results in cellular damage, impaired glucose-stimulated insulin secretion, and, eventually, cell death. Importantly, oxidative stress stimulates uncontrolled protein oxidation and destruction. This is further aggravated by a strong inflammatory component due to the innate immune response and formation of inflammatory mediators. We (Nissim, Strollo) previously demonstrated that most individuals with type 1 diabetes (T1D) as well as children at risk of T1D have circulating autoantibodies to oxidative post-translationally modified insulin (oxPTM-INS); we also reported that oxidized insulin epitopes stimulate both CD4+ and CD8+ T cells of individuals with T1D. While this work is suggestive that oxPTM-INS is a novel target of islet autoimmunity and a disease biomarker, we have not yet shown that oxPTM-INS is present in beta cells of human organ donors with T1D or at risk for T1D. This project aims to determine whether oxidized insulin is present in the beta cells, and to what extent, in the islets of organ donors with T1D or those with autoantibodies representing preclinical disease stages. We have developed antibody reagents to address this question. In further studies, circulating levels of oxPTM-INS neoepitopes may be also be exploited as biomarkers of beta cells stress and T1D. Antibodies against oxidized insulin may potentially be exploited to deliver therapeutic cargo to stressed beta cells, and hence the determination of the abundance of oxidized insulin in the pancreas of T1D and autoantibody-positive donors may support the development of nvoel therapeutics.
-

Clayton Mathews
University of Florida
Funded by: Breakthrough T1D
Islet resistance to T1DOur proposed studies will address a major gap in the understanding of T1D, namely, to identify the mechanisms of beta cell failure from a metabolic as well as an immunological perspective. This specific project has high potential to reveal significant, novel, and translatable information regarding beta cell dysfunction that will be of therapeutic benefit for individuals with both established T1D as well as persons with pre-diabetes. The goal is to identify components of the glucose sensing and insulin secretory machinery pathways that are altered during the natural history of disease progression in type 1 diabetes (T1D), and to characterize the metabolic and immunological signals responsible for the functional loss in beta cell activity that are specifically associated with this disease. This aim will be achieved by performing two lines of experimentation. First, gene expression arrays will be utilized to analyze islet RNA isolated from pancreatic sections of nPOD cases (representative from the natural history of T1D, alongside of appropriate matched controls) following laser capture microdissection (LCM). These analyses will be undertaken to determine the specific gene expression changes that result in beta cell dysfunction, as well as to identify the immunological signals which are responsible for beta cell failure in T1D. In the second we will use primary human islets obtained from cadaveric donors with T1D as well as non-diabetic subjects, to confirm and extend our associations of phenotypes reflective of beta cell dysfunction. Also, using extended in vitro culture systems to allow for a recovery of beta cell insulin secretory function, followed by expression analysis, we will identify changes in signaling that are reflective of both cellular damage and recovery. These experiments are posed to test the overall hypothesis that that following the appearance of autoantibodies, inflammatory signals act to increase islet inflammation and negatively impact beta cells through downregulation of genes essential for glucose sensing and insulin secretion. Based on previously published work, we expect that the effects of these signals are reversible. Elucidating the immune signals that induce beta cell dysfunction would have a major impact on future treatment strategies as they should provide a means to revive the endogenous beta cell mass.
-
Clayton Mathews & Martha Campbell-Thompson
University of Florida
Funded by: NIH-NIAID
Investigating the role of iron metabolism in beta cell function and the immunopathology of type 1 diabetesInsulin release from beta cells depends on ATP energy g enerated by mitochondrial . The electron transport chain is the primary group of proteins that make ATP energy and they require iron to do so. Consequently, iron is essential for insulin release, and iron deficiency inhibits beta cell function. Conversely, excess cellular iron leads to an incre ase in reactive oxygen species (ROS), which can damage cells and cause dysfunction . Evidence indicates that both R OS and mitochondrial dysfunction occur during diabetes progression . B eta cells have low antioxidant capacity making them susceptible to ROS from iron overload. Iron overload can cause mitochondrial fragmentation and dysfunction . Indeed, diseases of iron overload such as hereditary hemochromatosis and Friedreich’s ataxia have an associated risk of diabetes in the absence of autoimmu nity . I nflammation alters iron metabolism by promoting sequestration within cells. Prolonged inflammation can cause anemia of inflammation , which has been reported in patients with T1D, where serum iron levels are too low to promote the differentiation of red blood cells . An inflammatory signaling molecule called interferon gamma (IFNy) is increased in in dividuals who develop diabetes long before the onset of disease . IFNy has been shown to increase cellular iro n . Taken together, these data demonstrate the importan ce of understand ing iron metabolism under basal conditions and dur ing T1D disease progression in beta cells. Importantly, iron metabolism pathways may serve as targets for intervention for the treatment of diabetes. -

Raghavendra Mirmira & Teresa Mastracci
Indiana University School of Medicine
Funded by: Breakthrough T1D
Deoxyhypusine synthase: a novel target for beta cell protectionThe cellular processes giving rise to type 1 diabetes (T1D) and type 2 diabetes (T2D) involve the activation of inflammation, which leads to the eventual death of islet β cells. An urgent priority in diabetes research is the discovery of biomarkers (simple blood tests) that can assist in the identification of persons at-risk for disease for the purposes of early therapy. Recently, our laboratory has been investigating the involvement of a specifically altered form of the protein eIF5A (known as eIF5AHyp) in the progression of diabetes in mice. In previously published studies, we showed that eIF5AHyp is responsible for the production of a subset of inflammatory proteins in β cells and immune cells of the mouse. However, the specific types of cells that exhibit eIF5AHyp in human diabetes have never been examined, largely because specific reagents for tissue analysis have heretofore been unavailable. The purpose of this study is to determine the cell-type distribution of eIF5AHyp in the human pancreas and spleen from persons with T1D, T2D and controls. The results of this study will uncover whether the accumulation of eIF5AHyp-expressing cells are a marker for diabetic disease in the pancreas.
-

Feroz Papa & Mark Atkinson
UCSF
Validating KIRAs as β-cell-sparing drugs in islets from human patients with T1DOur lab has been focusing for many years on the intrinsic stress experienced by pancreatic islet β-cells, which (even in healthy states) live under a strict metabolic mandate to continuously produce and secrete insulin at intensely high rates that predispose these professional secretory cells to secretory exhaustion. β-cells evolved a high-capacity endoplasmic reticulum (ER) organelle in which the insulin precursor, proinsulin, undergoes initial oxidative protein folding and structural maturation. However, whenever secretory demand outpaces folding capacity, unfolded proteins begin to accumulate in the ER—a condition called “ER stress”.
ER stress activates an intracellular signaling pathway called the unfolded protein response (UPR) that augments ER chaperones and protein-modifying activities by triggering adaptive (‘A’)-UPR transcriptional and translational programs. But if ER stress remains unresolved, the adaptive UPR outputs wane and are replaced by “terminal” (‘T’)-UPR programs that actively promote loss of differentiated cell identity, cause sterile inflammation and senescence, and ultimately trigger apoptosis. Mounting evidence shows that high ER stress/and consequent A>T-UPR conversion causes β-cell apoptosis, leading to diabetes in mouse models of T1D. However, it remains unclear whether human islets also demonstrate an A- to T-UPR switch during progression to T1D.
-

Edward Phelps
University of Florida
Funded by: Breakthrough T1D
Mechanisms and dynamics of GAD and GABA in human beta cellsIn humans, GAD65 is one of the first targets of autoimmunity and 70-80% of human diabetics have antibodies against this protein. In late-onset patients, circulating GAD65 autoantibodies may even be more predicative of future disease onset than antibodies to insulin. The early appearance of GAD65 antibodies may hold a clue that GAD65 is implicated during the earliest phases of the disease, which makes it an attractive target for interventional therapies to prevent clinical progression. One of the peculiarities of GAD is that this protein is expressed in two isoforms, and only the lighter weight form, GAD65 is immunogenic, while the heavier one, GAD67, is not. Mice predominantly express GAD67 in their beta cells, while rats express both GAD65 and GAD67 and humans only express GAD65. Because the majority of diabetes research is conducted on NOD mice where GAD65 is not well expressed in beta cells, the central importance of GAD65 in human diabetes may be overlooked in NOD mouse research. We are studying GAD65 in human tissues to better understand how this protein contributes to T1D. In addition, we have an interest in the role of gama-aminobutyric acid (GABA), the enzymatic product of GAD. GABA is a potent neurotransmitter that is strongly and selectively synthesized in neurons and pancreatic islets. Beta cells secrete GABA as a paracrine and autocrine signal to help regulate hormone secretion. GABA is also a negative regulator of immunity and helps shape the islet local immune microenvironment.
-

Al Powers, Marcela Brissova and Rachana Haliyur
Vanderbilt University
Funded by: Breakthrough T1D and NIH
Pancreatic islet biology and vascularizationOur group is interested in pancreatic islet development and function and how beta cells are affected in diabetes. Critical discoveries from nPOD samples and resources are changing our understanding of type 1 diabetes. We are using or hope to use nPOD samples for the following studies:
1) Define and describe the normal human pancreas and islet over the human lifespan. In terms of normal pancreatic islet development, we are studying the fetal human pancreas, the juvenile human pancreas (less than age 18), and the adult human pancreas. We are interested in how islet morphology, gene expression, and function change as humans age and adapt to different physiologic conditions such as puberty and insulin resistance. In addition to studying islet gene expression (hormones, key islet-enriched transcription factors, and differentiation markers), we are also studying the innervation and vascularization of islets. Islets are richly innervated and vascularized and we are interested in interactions between these processes and islet cell function.
2) Describe the features of insulin+ cells remaining in the pancreas of individuals with Type 1 diabetes (T1D). We seek to understand the functional characteristics, and molecular signatures of the insulin+ cells remaining in the pancreas of individuals with T1D. Little is known about the remaining insulin+ cells and their functional status, or how those cells have survived in the inhospitable metabolic and immunologic environment. It is not known whether these insulin+ cells are ‘normal’ β cells.
3) Describe the features of islets and beta cells in the pancreas of individuals with cystic fibrosis-related diabetes (CFRD). This form of diabetes is becoming increasingly common, but the pathogenesis is incompletely understood. By studying samples in the nPOD repository, we hope to determine how these beta cells compare to normal beta cells.
We are using or hope to use nPOD samples from both the control and diabetic pancreas. We will combine information from nPOD samples with our results from samples from human pancreatic tissues that our group has recently collected (fetal and juvenile). We will examine these tissues by immunohistochemistry and other methods such as tissue imaging mass spectrometry. We will use antibodies that are widely available and also new antibodies that are being developed by investigators in the Beta Cell Biology Consortium.
July 11, 2014
-

Boone Prentice & Clive Wasserfall
University of Florida
Evaluation of Fatty Acid Levels in Human Pancreatic Tissue using Imaging Mass SpectrometryAlterations in metabolism are known to play profound roles inthe health ofindividuals with type 1 diabetes (T1D). These alterations lead to disruptions in cell structure, signaling, and energy homeostasis. While most studies focus on identifying genetic or protein signatures of disease, the metabolic profile is a far more dynamic, sensitive, and rapidly-responding molecular measure of phenotype. Free fatty acids (FFAs) in particular represent a class of metabolites known to play key roles in protective activities such as preventing beta-cell apoptosis, regulating plasma glucose levels, and enhancing insulin sensitivity as well as destructive activities such as exacerbating insulin resistance and producing lipotoxic conditions conducive to apoptosis. The nature of these roles is in part determined by the concentration of the FFAs as well as their molecular identity. Unambiguous detection and identification of these metabolites by conventional, antibody-based fluorescent imaging approaches is not feasible. Instead, we will leverage chromatography-tandem mass spectrometry (LC-MS/MS) to analyze homogenized tissues. However, LC-MS/MS lacksa spatial component to the measurement and does not provide for the direct in situmeasurement of metabolites in intact tissue sections. Detecting the spatial distributionand diversity of FFAs directly in pancreatic tissue, before these metabolites have been diluted in the bloodstream, would provide an unprecedented view of T1D metabolism. Herein, we propose to use label-free imaging mass spectrometry to enable the multiplexed and facile discrimination of metabolites in human pancreas tissue sections. We will analyze and quantify differences in the expression of free fatty acids between unaffected and T1D pancreases. Our preliminary data suggest that multiple genes involved in fatty acid biosynthesis are significantly upregulated in T1D tissue compared to unaffected pancreases. The generation of molecular maps by imaging mass spectrometry will determine whether the metabolic phenotypes agree with the genetic profiles of these individuals.
-

Chaitra Rao & Emily Sims
Indiana University School of Medicine
Characterizing the association of PD-L1 splice variants in type 1 diabetesType 1 diabetes (T1D) exhibits significant heterogeneity in disease progression, driven in part by variable immune-mediated destruction of insulin-producing β cells. This project focuses on Programmed Death-Ligand 1 (PD-L1), a critical immune checkpoint protein that protects β cells by suppressing autoimmune responses via interaction with PD-1 on immune cells [1]. β cells also release PD-L1 via extracellular vesicles (EVs), which may regulate immune activity beyond the islet microenvironment [2].
Our previous work revealed that exposure to interferons—a hallmark of the inflamed islet microenvironment—upregulates PD-L1 on both β cells and their EVs [2]. Using nPOD slice perifusates, we observed heterogenous elevations in EV PD-L1 levels isolated from pancreatic slice perfusates of T1D donors compared to controls. Within this group, EV PD-L1 correlated with preserved insulin secretion in situ, suggesting its potential as a biomarker for residual β cell function. However, mechanisms underlying variability in EV PD-L1 across individuals remain unclear.
Our preliminary data indicate that interferon exposure also induces an alternative splicing variant of PD-L1 lacking exon 3 (PD-L1Δ3). This variant is poorly incorporated into EVs and may reduce the protective PD-L1 cargo in these vesicles. We hypothesize that increased β-cell production of PD-L1Δ3 in some individuals impairs EV PD-L1 trafficking, leading to variable immune protection and increased β-cell vulnerability.
To investigate this, we will analyze panceatic sections using advanced single-molecule in situ hybridizaton (BaseScope assay) to detect PD-L1Δ3 variant in β cells from donors with, at-risk, and without T1D. By correlating these findings with existing EV PD-L1 data from perifusates, we aim to understand how alternative splicing alters PD-L1 trafficking and impacts β-cell survival. This research could uncover novel biomarkers and therapeutic targets to preserve β-cell function and slow T1D progression.
-

Sarah Richardson, Irina Stefana, John Todd & Noel Morgan
University of Exeter Medical School
Funded by: Breakthrough T1D, MRC and Wellcome
Investigating the role of aggregation-prone proteins in pancreatic beta cell function and healthAlmost 20% of the chromosomal regions associated with the risk of developing of Type 1 diabetes (T1D) identified to date,have no identified candidate causal gene. Often, this is because the genes in those regions have no known roles in the immune system, reflecting the classical view that T1D is caused by the autoimmune destruction of pancreatic islet β cells.One possibility is that the causal gene(s) in these regions are not acting in the immune system but rather on the target tissue (the βcells)or in both. Our ongoing analyses show that >150 genes in over 60 T1D risk regions are expressed in β cells and that several T1D-candidate variants colocalize to regulatory motifs active specifically in human islets and not in immune cells.One region that may act via βcells, rather than the immune system,to predispose to T1D is the chromosomal region 17q21.31. The same genetic signal in region 17q21.31 is also associated with increased 2-hr glucose levels following an oral glucose tolerance test and, importantly, with increased plasma proinsulin levels, thus strengthening the rationale that the association in this region might act via β cells. To date, this region has been extensively studied for its association with neurodegenerative diseases, where highly disease-penetrant mutations have provided evidence that MAPT, encoding for the microtubule-associated protein Tau, is the causal gene. Classically thought of as a neuronal protein, Tau is also expressed in several peripheral tissues, including pancreatic islet βcells.We are currently investigating the role of Tau in β-cellfunction and survival under diabetes-relevant stresses.Using nPOD samples, we aim to examine the expression and subcellular localization of Tau and its pathological, hyperphosphorylated formin human pancreatic β cells from donors of different ages and diabetes status
-

Teresa Rodriguez-Calvo
Helmholtz Zentrum München
ORIGINAL: Proinsulin processing and presentation in human type 1 diabetes: a beta cell perspective ADDENDUM: Proinsulin processing and presentation in human type 1 diabetes: a beta cell perspective –T cell perspectiveORIGINAL: Type 1 diabetes (T1D) is an autoimmune disease in which clinical symptoms arise as a result of insulin deficiency. While genetic and environmental factors contribute to the disease, in recent years it has become increasingly evident that beta cells might be contributing to their own destruction and might have an active role in type 1 diabetes development. Insulin production occurs thanks to processing enzymes, which transform the precursor of insulin (proinsulin) into insulin. Our hypothesis is that there might be a defect in the activity of these enzymes that would cause the accumulation of large amounts of proinsulin (which is not being transformed to insulin) and might also introduce insulin processing errors. These errors would lead to protein modifications that would be recognized by the immune system. Therefore, our objective is to investigate the mechanisms involved in proinsulin and insulin synthesis and the enzymes that process them. Consequently, we propose to study the biological processes that accompany insulin synthesis from a beta cell perspective, providing a more comprehensive understanding of the pathogenesis of type 1 diabetes. Ultimately, this work should help to develop novel therapeutic approaches targeting beta cell dysfunction possibly in combination with antigen-specific therapies.
ADDENDUM: Type 1 diabetes is an autoimmune disease in which clinical symptoms arise as a result of insulin deficiency. While genetic and environmental factors contribute to the disease, in recent years it has become increasingly evident that beta cells might be contributing to their own destruction and might have an active role in type 1 diabetes development. Insulin production occurs thanks to processing enzymes, which transform the precursor of insulin (proinsulin) into insulin. Our hypothesis is that there might be a defect in the activity of these enzymes that would cause the accumulation of large amounts of proinsulin (which is not being transformed to insulin) and might also introduce insulin processing errors. These errors would lead to protein modifications that would be recognized by the immune system. Therefore, our objective is to investigate the mechanisms involved in proinsulin and insulin synthesis, the enzymes that process them and which protein fragments (modified or not) are recognized by lymphocytes, causing their activation and leading to beta cell destruction. Consequently, we propose to study the biological processes that accompany insulin synthesis not only from the beta cell perspective but also from the side of the immune system, providing a more comprehensive understanding of the pathogenesis of type 1 diabetes. Ultimately, this work should help to develop novel therapeutic approaches targeting beta cell dysfunction possibly in combination with antigen specific therapies.
-

Teresa Rodriguez-Calvo
Helmholtz Zentrum München
The insulin secretory granule components and its immune recognition in type 1 diabetesOur parent project studies the intracellular accumulation and molecular interaction of proinsulin, insulin and the proinsulin processing enzymes PC1/3, PC2 and CPE, which may be dysregulated during the development of the disease and lead to processing errors. Our interest was focused on proinsulin and insulin, as these are the most abundant molecules of the insulin granule, and their role in disease progression has been highlighted in several studies but has not been systematically investigated. However, other molecules inside the insulin granule, such as chromogranins, secretogranins, and islet amyloid polypeptide (IAPP) might undergo similar processing errors. These may contribute to beta cell demise, thereby leading to their recognition by the immune system. Here, we propose to study the expression and accumulation of insulin granule proteins other than insulin and proinsulin. By approaching this from the perspective of the beta cell, we will strive to provide a more comprehensive understanding of the pathogenesis of type 1 diabetes. Ultimately, this work should help to develop novel therapeutic approaches targeting beta cell dysfunction possibly in combination with antigen specific therapies.
-

Teresa Rodriguez-Calvo & Aida Martinez-Sanchez
Helmholtz Diabetes Center & Imperial College London
Funded by: Steve Morgan Fouundation
Exploring miRNAs heterogeneity within pancreatic islet cells’ subpopulations in the context of T1DType 1 diabetes (T1D) happens when the immune system attacks and destroys the insulin-producing beta- cells in the pancreas, which are contained in the islets of Largenhans. Research has shown that beta-cells within islets are not all identical: some differ from others in the genes they use and in how well they work. We think that these differences, referred to as “heterogeneity”, affect how fast these cells are lost in T1D by making some beta- cells more susceptible to destruction by the immune system. Scientists are growing islets in the lab from stem cells (SC- Islets), hoping that these could be transplanted to cure T1D. We want to find out whether these differences between beta-cells are fully represented in SC-islets, as this may affect how well SC-islets work. For this, we need to better understand the heterogeneity of beta-cells in the islets of people with and without T1D, and whether it is similar to healthy SC-islets, and how does it change in inflammatory conditions. We are interested in a group of very small molecules called microRNAs. MicroRNAs help control which proteins a cell makes and are important for beta- cell development and function. However, no one has yet looked properly at whether microRNAs vary between different beta-cells within islets of Langerhans in the human pancreas during T1D. The project has two main questions: – Do miRNAs behave differently in different subtypes of beta-cells in people living with T1D compared to those without it, and during disease progression? – Are miRNAs different between subtypes of beta-cells in SC-islets compared with human islets? As part of aim 1, we will use pancreas samples from organ donors without diabetes, donors at risk of T1D , and donors with T1D. Using advanced imaging methods, we will analyze selected miRNAs directly in pancreas sections and examine which cells contain them, where they are located, and how they change during disease. We will also study how these patterns relate to insulin expression and the presence of immune cells. As part of aim 2, these images will be compared with similar stainings of SC-islets. Understanding these differences will help us understand why some beta- cells may be more vulnerable than others in T1D. Thus, this work will help us identify opportunities to prevent or decelerate beta-cell destruction in T1D or improve lab-made beta-cells for transplantation. -

Holger Russ
University of Colorado
Evaluating senescence induction in human beta cells upon transplantationWe are interested in cell replacement therapy as a potential practical cure for patients suffering from diabetes. Using stem cell derived beta cell clusters and cadaveric human islets transplantation in preclinical animal models we find distinct changes to the beta cell phenotype. This project will test if our observations hold true in islet recipients. We will evaluate if the changes observed using model systems are also present in tissue sections from donors that previously received islet transplants.
We find a small subpopulation of stem cell derived beta cells (sBC) marked by CD9. We found previously that CD9 marks beta cell subpopulations that are less functional in cadaveric islets preps (Dorrell et al. Nature com, 2016). CD9 cell % is up in T2D. When looking closely, part of the CD9 population in both sBC and cadaveric islets exhibits a senescent and senescence associated secretory phenotype. When we look at transplanted sBC we see a huge increase in CD9 cells (and senescence markers). We are currently testing if CD9 cells exhibit greater immunogenicity. We believe our findings have important implications to current efforts aimed at providing a practical cure via cell replacement therapy to diabetes patients.
-

Albert Salehi
Lund University
VDAC1 a Common Denominator of Pancreatic β-cell and Vascular Endothelial Cell Apoptosis in T1D and its Organ ComplicationsImpaired insulin secretion from the β-cells characterizes both type 2 and the initial phase of type 1 diabetes (T1D). Although the two sub-forms have different causes, we postulate that the rapidly developing β-cell defect and death in T1D as well as the slowly progressing β-cell dysfunction associated with T2D share a common cellular mechanism.
Long-term elevated plasma glucose (glucotoxicity) as well as presence of inflammatory cytokines in the pancreatic islets contribute to β-cell demise in both forms of diabetes. In the initial phase of T1D, islet cell antibodies are detected, while insulin secretion is attenuated but not abolished. Existing therapies are not preventing the progression of either T1D or T2D. We have recently shown that long-term exposure of pancreatic β-cells to high glucose (glucotoxicity) as well as β-cells isolated from T2D cadaveric organ donors show increased expression of the mitochondrial voltage-dependent anion channel-1 (VDAC1). This leads toVDAC1 miss-targeting (translocation) to the cell surface. Our preliminary data showed that inflammatory cytokines also increases VDAC1, which might contribute to the loss of β-cells in T1D. VDAC1 overexpression leads to its oligomerization, which participates in cell death. In preliminary results, we also observe upregulation of VDAC1 in a rat model of T1D. Since VDAC1 is a mitochondrial ATP transporter, its translocation to the cell surface is associated with ATP loss from the β-cells, leading to energy depletion. Prevention of ATP loss withVDAC1 inhibitors restores β-cell ATP levels and insulin secretion in T2D islets. Daily injections of a VDAC1 inhibitor prevent the onset of hyperglycemia in db/db mice, a T2D model. Based on the effects of cytokines, we want to extend our investigations to include T1D. To this end, we will studyVDAC1 protein expression and co-localization with other β-cell markers on pancreatic sections from T1D organ donors. We thus envision that novel therapeutics based on VDAC1 inhibitors i.e. newly developed small molecules or a specific VDAC1 antibody, may act either as complements to existing therapies, or as novel stand-alone treatments for T2D as well as prevention of the β-cell loss in T1D.
-

Charmaine Simeonovic, Christopher Parish, J. Dennis Wilson & Andrew Ziolowski
Australian National University
Funded by: Breakthrough T1D
Heparan sulfate levels mark the health status of human islet β-cellsHeparan sulfate (HS) is a sulfated glycosaminoglycan that consists of repeating disaccharides (composed of uronic acid and glucosamine), forming a linear polysaccharide. The HS chains are assembled onto the core proteins of heparan sulfate proteoglycans (HSPGs). We recently reported that HS is localized in the peri-islet basement membrane (BM) in mice and is intensely expressed and widely distributed throughout the intra-islet cell mass, being localized selectively inside insulin-producing beta cells. This intracellular distribution of HS is unique for beta cells, since normally HS is localized extracellularly in BMs and the extracellular matrix (ECM). Using in vitro studies, we have discovered that isolated mouse beta cells need intracellular HS for their survival and protection against oxidative damage. During T1D in NOD/Lt mice, HS is progressively lost from islet beta cells, due to the local production of heparanase (a HS-degrading enzyme) by insulitis leukocytes, resulting in beta cell death. Using nPOD human pancreas specimens, we have demonstrated that normal human islets also express high levels of intracellular HS in their beta cells. In parallel, our immunohistochemical analyses of the distribution of HSPG core proteins revealed strong intra-islet staining for collagen type XVIII and syndecan-1 HSPG core proteins. These findings provide support for the high levels of HS in human beta cells in situ. Like T1D in NOD/Lt mice and in contrast to normal human pancreas, nPOD pancreas specimens from donors early after T1D-onset (≤ 8 years post-onset) have shown selective loss of HS but not HSPG core proteins in remaining insulin-positive islet beta cells. In fact the HS content of T1D islets was only 10% of normal controls. In addition, peri-islet insulitis mononuclear cells in the T1D pancreas specimens strongly expressed heparanase. Our findings strongly support the testing of dual activity heparanase inhibitors/ HS replacers as new therapeutics for preventing T1D progression in new onset T1D individuals.
-

Emily Sims & Teresa Rodriguez-Calvo
Indiana University School of Medicine
Defining the link between Islet prohormone accumulation and secretion in type 1 diabetesAn improved understanding of the pathophysiologic mechanisms contributing to insulin deficiency in type 1 diabetes (T1D) is crucial to optimize approaches to disease prevention and treatment. Multiple groups have described increases in beta cell prohormone s in individuals at different stages of T1D development, both at the level of the islet and in circulation. These findings have highlighted the concept that dysfunctional beta cell prohormone processing may contribute to insulin deficiency and T1D progress ion. Because of historical limitations of access to human pancreatic tissue, prior human analyses have been unable to combine analysis of prohormone secretory dynamics with tissue – level phenotyping of prohormone expression and processing. The introduction of nPOD and other like programs, alongside innovative technologies like the tissue slice effort (the subject of this RFA), is markedly changing this situation. For this proposal, our overarching hypothesis is that dysfunctional prohormone processing at th e level of the islet will be correlated with altered islet prohormone release. Specifically, we predict that, relative to non – diabetic control donors, islets from donors with T1D will exhibit: 1) increased basal release of islet proinsulin and pro – islet am yloid polypeptide (proIAPP) relative to mature insulin and IAPP, 2) an abrogated increase in prohormone release under stimulated conditions, and that 3) these functional secretory abnormalities will correlate with islet prohormone accumulation. To test thi s, we propose analysis of islet prohormone secretion relative to mature hormone secretion from slices from nondiabetic donors and donors with T1D. Histologic analyses of proinsulin, proIAPP, and insulin and amyloid in slice sections will be performed. The se results will determine if islet prohormone expression and processing is linked to altered hormone secretion, yielding critical insight into association of altered prohormone processing and release with insulin deficiency in T1D. Additional exploratory m echanistic studies will test EV content using the remaining perifusate samples already at our possession at IU, using the Exoview platform, with quantification of PD – L1+ and LC3+ positive EVs (absolute and as a percentage of total EVs) as well as bead – based pulldown and permeablization to identify proinsulin+ EVs. These results will determine if heterogeneity islet proinsulin secret ion is linked to altered EV composition, yielding critical insight into association of altered prohormone processing and release with insulin deficiency in T1D. -

Jason Spaeth
Indiana University School of Medicine
Funded by: NIH/NIDDK and Indiana University School of Medicine
Transcription factor:coregulator interactions are compromised in islet cells under diabetogenic conditionsBeta cell dysfunction occurs in the early stages of type 1 (T1D) and type 2 diabetes (T2D) development. Strategies to improve outcomes for the mounting number of diabetic patients requires understanding the complex programs that coordinate a proper insulin release in response to changing blood glucose levels and how these mechanisms falter in diabetogenic settings. It has been proposed that expression and activity of islet enriched transcription factors (TFs) are altered in beta cells from diabetic individuals. We propose that interactions between TFs and their recruited transcriptional coregulators are negatively impacted during diabetes development and detrimentally alter gene expression programs essential for normal beta cell function. The Spaeth lab and others have demonstrated that Pdx1 interacting coregulators such as the Swi/Snf chromatin remodeling complex and the Nucleosome Remodeling and Deacetylase (NuRD) complex positively contribute to normal beta cell function in vitro and in vivo. Preliminary data suggests that beta cells from animals under high fat diet(which mimic human metabolic syndrome) and non-obese diabetic (T1D animal model)have reduced interactions between Pdx1:coregulators. We will utilize nPOD samples to evaluate if interactions between Pdx1 and these important coregulators are altered in primary human tissue from donors with multiple autoantibodies, overt T1D as well as donors that diagnosed as T2D.
-

Roland Stein
Vanderbilt University
Funded by: Breakthrough T1D and NIH
Lipid droplet distribution study in human pancreasIn collaboration with Dr. Alvin Powers’ at Vanderbilt University, we showed by electron microscopy (EM) that transplanted human islets have a much higher level of lipid droplets (LDs) than transplanted mouse islets (1). Moreover, we found that the number of LDs in transplanted human islets was increased in high fat diet (HFD) fed mouse recipients. We were intrigued by this observation, since we also observed that the high LD accumulation in human islets correlated with the unresponsiveness of the transplanted human islet β cells to the proliferation and insulin secretion signals observed in mouse islets (e.g. HFD & hyperglycemia (1)). Consequently, we extended this analysis by using both EM and the BODIPY 493/503 dye (i.e. a green fluorescent dye with hydrophobic properties that specifically stains neutral lipids and other lipophilic compounds) to analyze the LD content in intact human primary pancreas tissue sections. BODIPY 493/503 has the advantage over EM of allowing visualization of LD content/redistribution within a larger pancreatic area. BODIPY 493/503+ LD signals were shown to be: 1) randomly distributed between the acinar and endocrine compartment in healthy human samples; 2) in the α- and β-like cells generated from human embryonic stem cells; and strikingly, 3) enriched within T2D islets and longstanding T1D residual α cells in relation to the acinar space. These findings suggested the pancreatic LD accumulation patterns could be a marker of dysfunctional endocrine cells, and were published in Diabetes with Dr. Powers’ group (2). Dr. Xin Tong, the lead investigator on this project, also received a postdoctoral fellowship supporting this project through Breakthrough T1D in 2019. We now propose in Aim 1 to use frozen OCT embedded slides to obtain insight into the effect of gender and BMI on pancreatic LD distribution. While low LD levels were previously found in healthy juvenile/young samples, we never tested T1D donors within this age range, which could have a novel LD enriched endotype. Consequently, we propose in Aim 2 to perform a pilot study exploring the pancreatic LD accumulation patterns in juvenile – young adult T1D donors (i.e. 10-20 years). Successful assessment of the LD accumulation pattern in these conditions will help determine the relationship of LDs to islet function, and guide the human islet selection process for transplantation therapy.
-

Ian Sweet & Christiane Hampe
University of Washington
Funded by: Helmsley Foundation
Pathogenic Action of Autoantibodies on Islet Function and Viability in Type 1 DiabetesType 1 Diabetes (T1D) is known to develop over time when autoimmune processes attack and destroy beta cells thereby reducing the capacity to secrete adequate amounts of insulin. The relatively gradual progression of the disease offers an opportunity for therapeutic intervention to slow or prevent destruction of beta cells before the disease has reached symptomatic stages. Systematic development of drugs will be greatly accelerated with identification of steps that mediate the progression of the disease. An intrinsic hallmark of T1D is the appearance of several autoantibodies directed against islet cell antigens, including insulin and the 65kDa isoform of glutamate decarboxylase (GAD65). We recently revealed pathogenic effects of autoantibodies on isolated rat islets (that like human islets do express GAD65). The paradigm-shifting results of our now published study demonstrated the ability of GAD65mAb to
i. penetrate and accumulate in beta cells,
ii. localize to various and distinct subcellular structures and
iii. affect insulin secretory function.
Accordingly, our overall goal is to establish that our exciting in vitro results showing an impact of GAD65 autoantibodies (GAD65Ab) on intracellular processes in pancreatic beta cells support a mechanism for mediating Type 1 Diabetes (T1D). As a first step, we will endeavor to show the presence and effects of GAD65Ab in human islets and in particular in human islets from patients with T1D. Therefore we are requesting pancreatic slides to search for the presence of antibodies in GAD-positive vs. control patients.
-

Farooq Syed & Sangeeta Dhawan
Arthur Riggs Diabetes & Metabolism Research Institute, Beckman Research Institute of City of Hope
Funded by: Breakthrough T1D/NIH
Role Integrated Stress Response Pathways in Health and DiseaseType 1 diabetes (T1D) development is characterized by insulitis, resulting in β – cell stress and progressive β – cell dysfunction and death. However, recent evidence suggest the role of pancreatic β cells in amplifying this immune response through the presentation of immunogenic peptides and secretion of cytokines and chemokines, indicating β cells may be complicit during the development of T1D. In this context, we reasoned that intrinsic β cell stress pathways such as ER – stress, oxidative stress and the integrated stress response (ISR) could play role immune cell activation and destruction of β cells . The ISR is a complex program of gene expression and translational control which is regulated through the selective phosphorylation of ⍺ subunit of the eukaryotic translational initiation factor (eIF2 ⍺ ). Chronic ac t ivation of ISR can le a d to defective RNA translation and generation of abberent proteins or peptides leading to β cell dysfunction or death. Furthermore, ISR influences RNA epitranscriptomic modifications, such as m6A and pseudouridine, which impact transcript stability and translation efficiency during cellular stress. Beyond RNA modifications, the chemicals regulating these processes can also modulate epigenetic mechanisms, including DNA methylation and chromatin remodeling, driving transcriptional reprogramming. Together, these interconnected processes shape cellular adaptation, survival, and disease progression in T1D. -

Gladys Teitelman
SUNY-Downstate Medical Center
Preventive Insulin Therapy for Type 2 Diabetes (Addendum: Pancreatic beta cell heterogeneity; Addendum: Heterogeneity of human beta cells)Two major pathophysiologic abnormalities underlie most cases of type 2 diabetes (T2D): insulin resistance and defects in pancreatic beta-cell function. These defects are initially compensated by an increase in insulin secretion and in the number of insulin secreting cells. However, with time, beta cell dysfunction and T2D develops, requiring intervention therapies. Eventually, there is significant beta cell loss and increased difficulty in the management of hyperglycemia.
It has been postulated that the increased rate of insulin synthesis induced by hyperglycemia could lead to beta cell failure. Insulin, initially synthesized as a larger polypeptide, is modified in the endoplasmic reticulum (ER) and post-translationally converted into insulin within the secretory/storage vesicles prior to secretion. Two proteases, the prohormone convertases (PC) PC2 and PC3/1, are responsible for this conversion. Deficiencies in insulin production could be due to defects in the processing of precursor insulin molecules in the ER and/or the secretory granules. Defects in the ER function unleash the unfolded protein response (UPR), which can be adaptive and recover normal cell function. Alternatively if the stress is chronic and severe, the UPR initiates cell death.
In pilot studies, using immunocytochemical techniques, we discovered that chronic pharmacological induction of insulin synthesis in normoglycemic white outbred CD-1 mice induced the expression, in all islets of 30% of mice treated (N=10), of a transcription factor that indicates severe ER stress. Significantly, islets of CHOP+ mice had a dramatic decrease in the levels of PC3/1 and PC2. Similar defects were found in beta cells of all diabetic C57BI6/Ks db/db mice examined.
The objective of this proposal is to ascertain the clinical relevance of our findings. We wish to determine whether increased demand for insulin synthesis in diabetic patients induce CHOP expression and/or abnormal low levels of the proprotein convertases PC3/1 and PC2 in pancreatic islets.
Addendum 1 Summary:
In this extension of the proposal we will test whether the heterogeneity of PC1/3 expression in human beta cells correlates with differences in the level of expression of mature insulin, proinsulin and of transcription factors involved in insulin synthesis, Second, whether there is a parallel between expression of proinsulin/mature insulin and that of proteins involved in insulin secretion. The third specific aim will evaluate the level of expression of proinsulin and of molecules involved in its processing in T2D islets. This analysis will allow ascertaining if the phenotypic variability in T2D reflects the presence, in each pancreas, of normal islets and of islets with different stages of beta cell dysfunction.
Addendum 2 Summary:
This proposal will test the hypothesis that beta cell death in Type 1 Diabetes occurs sequentiallyand is initiated with the immune mediated -ablation of the ProIN+PC1/3-cells. Comparison of celltype in pancreatic islets from autoantibody positive, newly diagnosed and long term T1D donors may reveal heterogeneity in response of the three cell types to the immunological attack.
-

Peter Thompson
University of Manitoba
Therapeutic targeting of senescent β cells in Type 1 DiabetesA growing body of evidence has implicated various forms of beta cell dysfunction in the pathogenesis of Type 1 Diabetes (T1D). Recently we discovered that during the natural history of T1D, a subpopulation of β cells become senescent. Senescence is a cell type-specific form of permanent growth arrest, which occurs during aging, tissue damage or stress and involves progressively acquired phenotypes including activation of a DNA damage response, upregulation of cyclin-dependent kinase inhibitors, an apoptosis resistance phenotype and a senescence-associated secretory phenotype (SASP). Using nPOD pancreas specimens from a small cohort of donors, we previously found that β cells expressing senescence markers indicating the DNA damage response, cyclin-dependent kinase activation and SASP accumulated in islets of autoantibody-positive (1 or 2 autoantibodies) and recent onset T1D donors (diagnosed ≥11-12 years of age, 0-6 years post-diagnosis) relative to control donors without T1D. Studies in the nonobese diabetic (NOD) model demonstrated that senescent β cells are pathologic and accelerate T1D development, as small molecule targeting of the apoptosis resistance phenotype of these cells prevents T1D. Therefore,ourprevious studies suggest that the apoptosis resistance phenotype of senescent β cells is a target for therapeutic intervention to delay or prevent T1D. However, the proteins that confer the apoptosis resistance phenotype in senescent human β cells are not known, therefore it remains to be determined whether senescent human β cells can be selectively targeted by small molecule therapy. The goals of this project are to define the apoptosis resistance phenotype in senescent human β cells that accumulate in autoantibody-positive and recent onset T1D donors and to evaluate the efficacy of small molecule inhibitors of apoptosis resistance in targeting senescent β cell in islets ex vivo. These studies will take us closer towards the clinical translation of anti-senescence therapies for T1D.
-

Peter Thorn
University of Queensland
Funded by: NHMRC and Diabetes Australia
Beta cell polarityInsulin secretion is principally regulated by the beta cell response to glucose. This secretory response is multiphasic with an initial burst of insulin secretion followed by sustained or increasing secretion. This complex timing of secretion is important for body function and is lost in type 2 diabetes. Beta cells normally function, and are normally regulated, within the islets of Langerhans which are composed of many hundreds of cells. However, due to technical limitations, beta cell function within islets is not well understood. To this end we have developed new methods that study single beta cells within an islet. Our new evidence shows that beta cells are structurally polarised and secretion of insulin is targeted to the vasculature.
-

John Todd
University of Oxford
Funded by: Helmsley Trust and Wellcome Trust
Quantitative proteomic profiling of human islets in healthy and diabetes associated pregnancyNormal pregnancy is associated with a rise in insulin secretion, which begins at placental implantation and increases progressively in parallel with placental growth. Following placenta delivery there is a sharp postpartum decline in insulin secretion. Gestational diabetes (GDM),the development of diabetes for the first time during pregnancy, is associated with pancreatic beta-cell dysfunction. Rodent models have been used to elucidate the biological alterations in beta-cells during normal pregnancy. However, these findings have not been investigated in human pregnancy and little is understood about the mechanisms that contribute to beta-cell dysfunction in women with GDM.
From mouse models, it is proposed that lactogenic hormones (prolactin [PRL] and human placental lactogen [hPL]) secreted by the placenta drive beta-cell expansion in normal pregnancy. Lactogenic hormones activate the beta-cell membrane prolactin receptor (PRLR) to increase downstream serotonin production. Serotonin may either increase or decrease beta-cell proliferation, in a receptor-dependent manner: rodent models have shown that activation of the5-hydroxytryptophan receptor 2B (5-HT2B)increases beta-cell proliferation,whilst5-hydroxytryptophan receptor 1d (5-HT1D)receptor activation decreases proliferation. At midgestation,Htr2bmRNA levels are increased in mice, which normalise at the end of gestation, whilstHtr1dexpression is increased at the end of gestation. It is proposed that it is this switch in serotonin receptor expression that contributes to the change in beta-cell mass during and following pregnancy. Preliminary data from our existing nPOD project illustrates that PRLRand5-HT1D receptor expression is increased in3rdtrimesterhuman beta cells in women with normal pregnancy but not in GDM. However, we have further found thatthe5-HT2Breceptor is not detected in normal human pregnancy or GDM at any stage during in gestation. Hence, much is still to be understood regarding pancreatic pathogenic mechanisms in GDM whilst, even in normal pregnancy, mouse models may not fully clarify pregnancy-associated changes in pancreatic biology.
In this study extension, we propose to study the proteomic profile of human islets from women with normal pregnancy and GDM. Towards this, we will isolate human pancreatic islets using laser capture microdissection (LCM) and perform proteomic analysis of the isolated tissue by quantitative liquid chromatography-tandem mass spectrometry (LC-MS/MS) to evaluate differences in islet protein expression and uncover pathways that are altered during normal and GDM pregnancy. This will improve our understanding of the changes in beta-cell biology in normal pregnancy, as well as GDM, and potentially provide valuable insights on whether they may be harnessed as beta-cell regenerative therapies for patients with diabetes.
-

Steven Zhang
Rutgers Cancer Institute of New Jersey
Role of Rab1A in Diabetes MellitusWe previously identified the Rab1A small GTPase as a conserved activator of mTORC1 in response to amino acid (AA) stimulation in yeast and mammalian cells. To better understand the physiological function of Rab1A- mTORC1 signaling in vivo, we genetically engineered conditional Rab1A knockout (Rab1A-KO) in mice. We found that tamoxifen-induced Rab1A knockout in young adult mice impairs insulin expression and beta cell proliferation, leading to hyperglycemia and glucose intolerance. Using beta-cell lines, we further found that Rab1A mediates AA signaling to regulate insulin transcription and beta cell proliferation through phosphorylation of the transcription factor Pdx1. Our results revealed a novel role of AA-Rab1A-mTORC1 signaling in controlling whole body’s glucose homeostasis in mammals. The objective of this study is to use human patient samples to obtain clinical evidence that Rab1A plays an important role in the progression of diabetes mellitus.
Beta Cell Development, Differentiation & Regeneration
-

Mark Atkinson
University of Florida
Funded by: NIH
Quantitative and qualitative evaluation of the endocrine & exocrine pancreas in type 1 diabetesIn the most widely recognized natural history model for type 1 diabetes, there is a presumption that all individuals begin life with the same number of beta cells. In combination with this, a consensus exists that the so-called “normal” adult pancreas possesses approximately one million islet cells. These concepts have drawn little attention or question over time as much of the research interest in terms of the disorder’s natural history has focused on genetic susceptibility and the trigger period, where the beta cells begin a process of linear loss. However, a limited body of largely unnoticed data draws question to the notions of a normal pancreas as well as the number of islets and/or beta-cells that may be resident, both in early life through adulthood, in humans.
We (in association with Dr. Martha Campbell Thompson and Dr. John Kaddis) recently published data (JAMA, 2012) involving nPOD tissues suggesting that the pancreas of individuals with type 1 diabetes is smaller, consistent with other studies over the previous two decades. But, until recently, the observation drew little investigator attention. Moving away from pancreatic size, the situation becomes even more intellectually diffuse when one considers quantification of islet and/or beta-cell numbers. In studies of pancreatic development in both humans and rodents, it appears that a wave of beta-cell death occurs as a normal physiological process following birth. The implications for this process may influence T1D’s pathogenesis in at least two ways. First is how it might influence T1D relates to the initiation of autoimmunity. A second notion as being of equivalent if not an even greater potential relates to the aforementioned concept of “100%”. We believe it conceivable that a major and yet highly unappreciated risk factor for T1D resides in the number of beta-cells present in early life. Taken collectively, these findings support a hypothesis that immune and perhaps non-autoimmune mechanisms (e.g., developmental, environmental, genetic, etc.) influence a loss of beta-cell mass as well as loss of pancreatic mass in T1D, possibly even before full-blown disease; thus, forming clear objectives for our studies.
Type 1 diabetes patients often exhibit exocrine insufficiency (REF), however, it remains unclear whether these alterations result from disrupted islet-acinar interactions secondary to the loss of functional β-cell mass or contribute directly to type 1 diabetes development. Thus, as a basis for a better understanding of pancreatic exocrine function, we propose to investigate the expression pattern of major digestive enzymes in the pancreas of nondiabetic, autoantibody positive without diabetes and T1 diabetes nPOD donors. This will be achieved by multiplex immunohistochemical staining for amylase, lipase and trypsinogen (amongst others) and by employing fresh tissue slicing technique to obtain functional readouts of pancreatic endocrine and exocrine cell function .
-

Anil Bhushan, Mark Atkinson, Todd Brusko & Bita Brodbar
University of California, San Francisco
Funded by: Breakthrough T1D
Epigenetic analysis of nPOD islet cellsType 1 Diabetes (T1D) is a complex autoimmune disease affecting more than 30 million people worldwide. T1D is caused by a combination of genetic and epigenetic factors leading to immune destruction of insulin-secreting islet cells. Although significant progress has recently been made in elucidating the genetics of T1D, the non-genetics component has remained poorly defined. Multiple lines of evidence including studies of migrant populations and monozygotic twin pair discordance for T1D underline the role of epigenetic factors in this disease. Thus, the characterization of epigenome is critical for the development of new T1D therapeutics and for appropriate staging of disease. DNA methylation is one of the main epigenetic modifications involved in human disease. Using epigenome-wide analysis such as reduced representation bisulfite sequencing (RRBS), we aim to identify unique differentially methylated regions (DMRs) within the genome that accumulate in PBMCs isolated from circulating blood, that may reflect the immune insult that occurs with T1D onset. Then we are interested to study the role of these hypo or hyper methylated regions in T1D.
-
Susan Bonner-Weir & Anmar Khadra
Joslin Diabetes Center
Funded by: Rumsey Funds
Origin of composite islet structure in humansIn the adult human pancreas the majority of islets have a clear non-random organization of mantle of non beta cells around beta cell cores, with larger islets usually having multiple such subunits (composite) whereas smaller islets resembled the classic rodent pattern. There is far more variability in islet composition both within each human pancreas and among different human pancreas than in rodent pancreas. The intrapancreatic variability in human pancreas raises important questions about how islets evolve and function throughout life and how this might relate to the pathogenesis of diabetes. We will examine the origin of the composite structure of the human islets by histological analysis of pancreas of donors from newborn to 25 years old. Determining the islet organization pattern (composite vs simple) with the distribution of islet size over the age range of expansion of the beta cell mass will provide data for mathematical modeling, which will predict the mechanism of formation of the composite islets.
-
Susan Bonner-Weir & Jens Hoiriis Nielsen
Joslin Diabetes Center
Mechanisms Involved in Expansion of Beta Cells in PregnancyPregnancy is associated with a marked increase in the number of pancreatic beta cells in response to the increased insulin demand. In rats and mice the growth of the beta cell mass is primarily due to proliferation of the existing beta cells resulting in enlargement of the islets of Langerhans. However, in a study of islets in human pregnancy (Butler et al 2010) the beta cell mass was found to be increased by numerous small islets and no sign of proliferation, suggesting that the growth of the beta call mass is due to neogenesis from putative progenitor/stem cells. In the proposed project we want to test the hypothesis that neogenesis rather than proliferation of beta cells is involved for the expansion of the beta cell mass in human pregnancy. This will be performed by immunochemical staining for prolactin receptors, Ngn-3 and Pdx-1 as markers of beta cell regeneration in pancreatic sections from pregnant women.
-

Alexandra Butler and Mark Atkinson
Royal College of Surgeons in Ireland - Bahrain
Determination of RFX3 and RFX6 expression in human fetal, neonatal, childhood and young adult pancreasThe human pancreas is a vital organ responsible for both endocrine and exocrine functions, playing a crucial role in glucose metabolism and digestion. It originates from the foregut endoderm, with the dorsal and ventral buds forming and eventually fusing t o develop into a mature pancreas. Key stages of pancreas development include specification, branching morphogenesis, and differentiation into endocrine and exocrine cell types ( Jennings et al., 2015 ) . Transcription factors play pivotal roles in pancreas de velopment. Some of the key transcription factors include : PDX1 (Pancreatic and Duodenal Homeobox 1): e ssential for early pancreas development and beta – cell differentiation (Offield et al., 1996 ) ; m aintains progenitor cell populations and is involved in ductal and endocrine differentiation (Seymour & Sander, 2011 ), NGN3 (Neurogenin 3): NGN3 m arks endocrine progenitors and is crucial for islet cell development (Rukstalis & Habener, 2009). NKX6.1: NKX6.1 Plays a key role in the maturation and function of beta cells (Sander & German, 1997 ) RFX3: RFX3 is a critical transcription factor involved in the development and function of the endocrine pancreas. It plays a significant role in regulating gene expression necessary for the growth of cilia in pancreatic endocrine cells in mice , which is essential for maintaining the cellular composition of islets of Langerhans, insulin production, and glucose tolerance. RFX3 is also crucial for the differentiation and function of mature β – cells and directly regulates the β – cell promoter of the glucokinase gene, a key enzyme in glucose metabolism in the mouse pancreas ([Ait – Lounis et al., 2007] [Ait – Lounis et al., 2010 ] ). While its role in mouse pancreatic development is less defined, its role in human pancreas development has never been explored before. RFX6: RFX6 is a crucial transcription factor involved in the development and function of the pancreas, particularly in the differentiation of insulin – producing β – cells and regulation of insulin production. It is essential for the proper differentiation of endocrine cells, as seen in conditions like Mitchell – Riley Syndrome, where mutations in RFX6 lead to impaired pancreatic development (Trott et al., 2020 ). RFX6 also influences insulin synthesis by regulating genes involved in mRNA translation (Cheng et al. , 2019 ) and mutations in this gene are linked to neonatal diabetes and other pancreatic dysfunctions (Concepcion et al., 2014 ) , however, its role in human exocrine and endocrine pancreas has ne ver been explored before. Transcription factors belonging to the regulatory factor X (RFX) transcription factors are involved in regulating genes critical for ciliary development and function, playing roles in organogenesis and cellular differentiation across various tissues. The exact role of RFX3 in pancreas development is less understood but is thought to involve the regulation of gene expression patterns crucial for pancreatic differentiation (Chung et al., 2020 ). In th is project , we are seeking to investigate the expression of RFX3 and RFX6 in human fetal , neonatal , childhood and young adult pancrea tic tissues to identify crucial novel factors of human pancreas development. -

Martha Campbell-Thompson, Alberto Pugliese, Clive Wasserfall & Mark Atkinson
University of Florida
ORIGINAL: Beta cell regeneration for Type 1 Diabetes ADDENDUM: Neuro-insular pathways regulating beta cell regenerationORIGINAL: Understanding basic human beta cell physiology is crucial to targeted therapies and prevention strategies for type 1 diabetes. Clinical signs in type 1 diabetes start when beta cell function and/or numbers are unable to maintain adequate blood glucose levels. After onset of type 1 diabetes, life-long insulin replacement therapy is needed. Factors regulating human beta cell regeneration are poorly understood. The purpose of these studies are to: 1) define pancreatic beta and alpha cell populations and how diabetes alters their function and numbers, 2) characterize potential autoimmune mediators of islet inflammation (insulitis) and resultant beta cell death, and 3) correlate lymphocytic populations in pancreatic and nonpancreatic lymph nodes and spleen between pre-diabetes and type 1 diabetic patients.
ADDENDUM: The goal of this addendum is to utilize large scale 3D human pancreas imaging methods for quantification of islet neuroendocrine cells and intra-pancreatic neurons and fibers. The autonomic neuro-insular network may be an unrecognized co-factor in the loss of functional beta cells during the progression to type 1 diabetes and following an islet transplantation. This proposal is expected to provide key data regarding neuronal pathways regulating beta cell regeneration.
-

Jan Czyzyk
University of Minnesota
Funded by: Breakthrough T1D
Expression of pancreatic Notch1 in type 1 diabetesPancreatic tissue is composed of two dominant cell types: small clusters of insulin producing cells called the islets of Langerhans and exocrine cells in the surrounding stroma. The endocrine and exocrine pancreatic tissues are fundamentally different in terms of anatomical organization, basic function and pathological conditions, and they are consequently believed to be largely independent from each other with respect to their biological roles. Our research is based on the premise that the endocrine and exocrine pancreas do not operate in isolation, but rather have a close functional relationship that influences islets both under normal and under diabetic conditions. Specifically, we propose that crosstalk between exocrine and endocrine pancreatic tissue is dependent on islet sensing signals that are generated in the surrounding tissue – a process we hypothesize is regulated by proteases. Although proteases, and the immune response to protease inhibitors, have been implicated in the pathogenesis of inflammation and autoimmunity, studies in our laboratory suggest that an immune response to the serpin protease inhibitors and the consequent increase in protease activity is unique in that it is protective rather than pathogenic during the diabetogenic destruction of pancreatic islets.
-

Juan Dominguez-Bendala, Giacomo Lanzoni & Luca Inverardi
University of Miami
Funded by: Diabetes Research Institute Foundation
Progenitors of the beta cell lineage in the pancreas and biliary tree of diabetic patientsStemming from a collaboration with Lola Reid (University of North Carolina), we have described stem and progenitor cells in niches of the human biliary tree and pancreatic ducts. These cells can mature into pancreatic endocrine cells, including functional insulin-producing cells. Our data suggest that bile ducts, peribiliary glands, pancreatic ducts and pancreatic duct glands may comprise a continuous ramification of cells that are related in maturational lineages. We have identified an extended network of stem cell pockets that branch out from the more naïve stem cells in the biliary tree to more and more mature cells in the pancreas. The presence of these progenitors in adults strongly suggests that the human pancreas has the potential to replenish lost cells in normal homeostasis and disease. Preliminary data obtained through our nPOD collaboration with Ivan Gerling (University of Tennessee) suggest that islets at the early stages of Type 1 Diabetes (T1D) may be undergoing regeneration or neogenesis. We are: (1) investigating the presence of pancreatic endocrine stem/progenitor cells in T1D and T1D Medalist cases; (2) testing if the stem/progenitor cell pool is affected by (or responsive to) the pathology and (3) studying whether these progenitors are subject to autoreactive aggression or not. We are also studying progenitor cells in transplanted pancreata in the nPOD-T study group. The overall aim of the project is to understand if regenerative processes are ongoing in T1D, to clarify the grade of maturation of beta cells in T1D, and to determine the feasibility of autologous approaches aimed at beta cell regeneration in T1D patients. Our team is open to new nPOD collaborations; we offer expertise and technologies in the following fields: stem cell markers, pancreatic islet development, beta cell maturation, immunology, imaging, laser-capture microdissection.
-

Juan Dominguez-Bendala
University of Miami
Funded by: NIH/NIDDK
Project 1: Human Pancreatic Slice-Based Studies of Regeneration and Exocrine–Endocrine Cross-Talk (In Vitro)The different cellular compartments of the pancreas have a high degree of plasticity, which, if harnessed, could have important therapeutic implications for beta-cell regeneration in type 1 and type 2 diabetes. This proposal focuses on high-resolution characterization of ductal progenitor-like cells (P2RY1+/ALK3+) and exocrine-endocrine communication in live human pancreatic slices. We will use dynamic single-cell RNA sequencing, live imaging/lineage tracing, spatial transcriptomics, and pharmacological perturbation with BMP pathway agonists and other candidate regenerative agents to define ductal remodeling, beta-cell neogenesis, and tissue-level signaling in human pancreatic tissue. This revised application clarifies that rare T1D and AAb+ slices will be used only for experiments requiring the diabetic or prediabetic tissue context, after technical optimization in non-diabetic control slices whenever possible. -
Juan Dominguez-Bendala
University of Miami
Funded by: NIH/NIDDK
Modeling Autoimmune β-Cell Destruction in Human Pancreatic Slices, SliceChip, and Slice-in-ACE PlatformsAutoimmune β-cell loss in T1D occurs in a three-dimensional tissue context that current cell culture systems fail to replicate. HPSs preserve native islet-exocrine relationships and tissue architecture, providing a unique human platform to observe immune damage in real time. This revised application focuses on building controlled models of autoimmune β-cell injury, not on requesting rare disease-context tissue for initial model development. In collaboration with the Pastori, Agarwal, Abdulreda, Nakayama, Panzer, and Pugliese teams, we will combine non-diabetic HPSs with HLA-compatible autoreactive TCR transductants and cytokine conditioning in three complementary systems: standard slice culture, SliceChip/vascular SliceChip, and Slice-in-ACE. The goal is to establish interpretable platforms that distinguish antigen-specific β-cell injury from allogeneic or nonspecific tissue damage. Future use of T1D or AAb+ slices would be considered only through a later addendum or nPOD-approved rotation after completion of the go/no-go milestones described below. -
Juan Dominguez-Bendala
University of Miami
Funded by: NIH/NIDDK
Human Pancreatic Slice-in-ACE Models for Regenerative StudiesThe developmental plasticity of the pancreas has been extensively studied. Leaving aside the ongoing debate about the relative contribution of neogenesis and endocrine cell self-duplication to islet regeneration in mice, the notion that progenitor cells can regenerate islets has been recently substantiated in human models. That this potential is intrinsic is evidenced by the observation that sorted human progenitors recapitulate complex, anatomically accurate pancreatic structures when transplanted into immunodeficient mice. An already substantial body of work also points at tissue stress and inflammation as critical drivers of this regenerative program –which would explain why ductal remodeling and intraductal endocrine cells are commonly observed during pancreatic injury and diabetes but seldom in the healthy organ. We have additionally established that BMP signaling stimulation accelerates this process, both in vitro and in vivo , opening the door to pharmacological approaches to induce β -cell regeneration in situ . Still, there remain many open questions. To put it plainly, nobody has ever seen regeneration unfold in real time in a live pancreas. We rely mostly on in vitro models (which show regeneration but may not accurately represent in vivo phenomena), inferences from static data (e.g., β -cells in apposition to ducts following pancreatic injury or pseudotime cell trajectory predictions), or the post-hoc interpretation of lineage tracing observations (which are often contradictory owing to technical and biological reasons). Our team has gone one step further by dissecting the process dynamically at the single-cell level using long-term cultured mouse and human pancreatic slices. However, for all the might of this organotypic model compared to other alternatives, they remain ultimately an in vitro system –and, as such, weighed down by caveats (chiefly lack of blood circulation) common to all such settings. These technological bottlenecks have led to a situation in which every finding can be either qualified or disputed based on the perceived limitations of any given model or the interpretation of their output, prompting intense academic debates about the nature and extent of pancreatic regeneration. We hypothesize that the transplantation of human pancreatic slices or sorted progenitor cells into the anterior chamber of the eye (ACE) of immunodeficient murine hosts will address the above shortcomings by enabling the longitudinal study of human progenitor cell potency and islet regeneration over many months. The efficient engraftment of human pancreatic tissue in this location affords us the possibility to delve into a living, functional, and fully vascularized representation of the pancreas in real time. While no model will ever be as complex and nuanced as the original subject, we contend that ours represents a first in the field: a section of the human pancreas in vivo in a completely controlled and accessible environment that permits visualization of (and intervention in) the dynamic growth of pre-existing islets and the appearance of neogenic β -cells over weeks and months . -

Farzad Ensi
University of Pittsburgh
Funded by: NIH and Children’s Hospital of Pittsburgh of UPMC Research Advisory Committee
Conversion of human acinar cells into insulin – producing cellsReprogramming of acinar cells toward functional β-like cells would offer an abundant and autologous source of insulin-producing cells. The current literature suggeststhat manipulation of genetic factors,in combination with cytokines or small molecules targeting specific pathways and the epigenetic machinery in humans,may eventually yield functional acinar-derived β-like cells. Our preliminary datashowthat treatment of adult mice with a compound that specifically inhibits the kinase activity of focal adhesion kinase (FAK) results in transdifferentiation of acinar cells into insulin-producing cells. Notably, the acinar-derived insulin producingcells infiltrate the pre-existing endocrine islets and are able to normalize the blood glucose levels.Here, we seek to determine whether FAK inhibition would convert human acinar cells into insulin-producing cells.
-

Kevan Herold & Mark Mamula
Yale University
Analysis of cellular proteins in β cells from patients with type 1 diabetes and healthyOur objective is to identify characteristics of β cells that survive immune attack. Our hypothesis is that
these residual cells express immune inhibitory molecules and/or decrease expression of diabetes
associated antigens. In addition, these cells may show changes consistent with dedifferentiated β cells.
Our studies of protein modifications have also shown that there are post-translational changes in
proteins produced by β cells during disease development. Finally, we have developed T cell library
assays that will enable us to identify novel antigens that may be found in islets during progression of
diabetes. Our objective is to determine whether β cells with these features can be found in the islets of
patients with T1D. -

Pedro Herrera
University of Geneva Medical School
Funded by: NIDDK and Breakthrough T1D
Cell conversion in the human pancreasThe overall goal of my proposal is to identify means for improving β-cell regeneration in the adult pancreas. We previously developed a transgenic model of inducible total or partial β-cell ablation (termed RIP-DTR). We have reported that in these mice there is spontaneous reconstitution of new β-cells from heterologous (i.e. non-β) cells after near-total β-cell loss.
The RIP-DTR model has revealed an unsuspected degree of cellular plasticity in the pancreas of juvenile and adult mice, including aged individuals, regarding the spontaneous inherent capacity of islet a-cells to switch to insulin production upon β-cell loss.
During the next years we want to address the following fundamental questions:
1. What is (are) the signal(s) driving a-cell reprogramming upon near-total β-cell ablation?
2. Can the a-to-β-cell conversion be fostered? Why only a small fraction of a-cells engages into conversion? What is the nature of the epigenetic modifications in reprogrammed a-cells?
3. Can human a–cells reprogram to insulin production?
4. What is the influence of ageing on islet cell plasticity?
5. Can other islet cells, i.e. besides a-cells, reprogram to insulin production?
6. Can in vivo a-cell conversion be facilitated by means of compounds mimicking the effect of instructive signals?
7. Are human islets endowed with cell plasticity capabilities? How diabetic conditions influence i) the capacity of human/mouse a-cells to reprogram, and ii) the maintenance of the β-cell phenotype?
-

Pia Leete & Joanna Boldison
University of Exeter Medical School
Funded by: RD Lawrence Fellowship Diabetes UK & Leona M. and Hary B. Helmsley Charitable Trust
The Heterogeneity of Type 1 Diabetes: An Islets StoryWhen a person is diagnosed with Type 1 diabetes (T1D), t he amount of beta – cell mass each individual lose s is highly variable despite their clinical need for insulin therapy. Attempts to detect robust markers in the blood that precisely describe disease activity in the islets are ongoing , but without a full understanding of the disease course in the pancreas, when attempting to target therapy to the organ , we are still operating par tially blindfolded. We have previously reported that two forms, or endotypes, of T1D are evident in the pancreas [1 – 4] . T hese differ according to the degree of beta – cell loss, the composition of the islet – specific immune infiltrate , and how beta cells process insulin . Additionally, we now have preliminary data that suggests the beta – c ells them se lves may vary between the two forms ) , even hint ing that successful beta – cell regeneration may be more likely in one form than the other . Our proposed study will expand on these investigations , and we will compare pancreatic protein expression profiles between people diagnosed with T1D at different ages, with different durations of disease and compare this to those without diabetes and with Type 2 diabetes (as a proxy for metabolic dysregulation) . We will leverage advanced histological and imaging techniques to analyse the presence of multiple critical proteins in granular detail in whole sections of pancreas . W e will then (b ased on several features of pancreatic T1D and beta – cell loss ) , construct a nuanced ‘pseudo’ timeline of disease progression in T1D to illustrate the life and loss of beta – cells in T1D and explo re if this is the same in all p eople, randomly different between people – or st ra tifiably different between gro ups of people living with T1D . With this information we will bett er understa nd if we should be looking for distin ct forms of T1D , and this may h elp scientists and trialist s underst and why some people re spond brilliantly in clini cal trials, whilst other do not. -
Heiko Lickert & Teresa Rodriguez-Calvo
Helmholtz Zentrum München
Identification of novel monoclonal antibodies directed against pancreatic endocrine cell types for in vitroand in vivo analysis and targetingDiabetes mellitus is a multifactorial disease characterized by progressive loss or dysfunction of the insulin-producing beta cells in the pancreas that leads to chronic hyperglycemia, systemic metabolic complications and multi-organ damages. Nowadays, no current treatments can stop or reverse the disease progression, apart bariatric surgery and human islet transplantation. Therefore, intensive efforts in the field of diabetes research are put into the development of novel therapeutic approaches.Regeneration of β-cells or replacement by β-like cells derived from stem cells hold great promise to stop or reverse disease progression. Unfortunately, the development of new treatment options is limited by our poor understanding of human pancreas organogenesis. The differentiation programs of all islet cells and the way in which islets are formed during pancreas development in their body niche, need to be fully understood1-3.Recently, many promising discoveries that could potentially lead to stem cell-derived functional human islets were made. However, the field currently still lacks of specific tools that can allow for enriching the endocrine progenitors during the differentiation process to improve efficiency and functionality. To date, there is only one beta cell marker available, which is already limited in that it doesn’t mark all beta cells and in early stages of life is not specific for beta cells4. To fill in those gaps, in a screen to identify novel monoclonal antibodies (mAbs) that can be used to isolate and/or target human pancreatic cells, we have identified several mAbs that specifically identify pancreatic endocrine, exocrine and endothelialcells. The mAbs produced have a great potential as: 1) tools to isolate pancreatic cells types and their different subtypes for in-depth characterization of pancreas organogenesis 2) markers to characterize pancreatic cell heterogeneity during diabetes progression 3) novel biomarkers of diabetes 4) as a method to enrich for endocrine progenitors during human stem cell differentiation for cell therapy 5) specific carriers for drug delivery or direct therapeutic tools.
-

Chloe Rackham & Sarah Richardson
University of Exeter
Funded by: Breakthrough T1D
Exploring an islet-protective role for native pancreatic mesenchymal stromal cells in health and in type 1 diabetesOur research aims to investigate how a group of adult stem cells, known as mesenchymal stem /stromal cells(MSCs), located in the pancreas may be involved in helping to protect people from developing type 1 diabetes(T1D). MSCs are found in most parts (tissues) of the body, including the pancreas, bone marrow, fat and kidney. When samples of these tissues are taken, MSCs can be isolated in the lab and when fed with appropriate nutrients will expand in number, so that we have enough MSCs that may be used therapeutically to treat inflammatory and autoimmune conditions, including T1D. Once isolated and expanded in this way we refer to these cells as exogenous MSCs, as opposed to endogenous/native MSCs that are found naturally in the tissue from which they were isolated.
Researchers in the diabetes field often refer to these exogenous MSCs as ‘Islet Helper’ cells. This is because experimental studies have shown that exogenous MSCs improve the ability of islet beta cells to produce insulin in response to increased sugar levels. Additionally, experimental studies have shown that exogenous MSCs can help to protect islet beta cells from immune attack and destruction. Clinical trials have shown that that exogenous bone marrow MSCs help to preserve the function of insulin producing islet beta cells in some individuals with newly diagnosed T1D, after infusion into the blood circulation.
Whilst we have established a lot about the function of exogenous MSCs, our understanding of endogenous/native MSCs located in the pancreas, with regards to their role in health and T1D is limited. We aim to determine whether the number, distribution and function of native pancreas MSCs is altered in T1D.We will determine whether native pancreas MSCs have a role in health to protect pancreatic islets from immune attack and destruction. These studies will provide scientific information on MSC-related changes inT1D, that we can target therapeutically to help delay or prevent the progression of diabetes. We will further establish whether there are differences in native pancreas MSC number, distribution and function in different subgroups of individuals with T1D, thus determining which subgroups are most likely to benefit from MSC-based therapeutic intervention.
-
Sarah Richardson & Xinpeng Dun
University of Exeter
Small endocrine objects – the key to endocrine mass expansion?Advances in 2D and 3D imaging techniques and technologies have enabled deeper interrogation of rare pancreas bioresources, highlighting significant gaps in our knowledge, with implications for the understanding of normal development and pancreatic disease. 3D volumetric imaging of the adult pancreas has revealed that 40 – 50% of the total endocrine objects within the organ consist of single beta cells or small clusters of beta cells. Despite their large numbers, these constitute a relatively small proportion of the total endocrine area, and they are not routinely assessed in histological, functional, transcriptional or proteomic studies. Virtually nothing is known about their role and function across the life course . We have now replicated the initial 3D observations by employing 2D image analysis of > 25 0 healthy and type 1 diabetes donor pancreata across different ages using AI – assisted pipelines that have enabled the study of > 45 0,000 endocrine objects (EOs) (Murrall et al, 2025 Science Advances) . Our results reveal a dramatic change in EO architecture and number in early development. With increasing donor age, EOs occupy an ever – decreasing proportion of the total endocrine area but still remain present in high number s into adulthood in subjects without diabetes. We carefully mapped the architectural changes within the pancreas that occur during the first few years of life and found that alterations in endocrine object size and architecture coincide with a dramatic exp ansion of the pancreas over the early years of life. As age increases, a marked decline in the proportional endocrine area (%) and endocrine object density occurs. This proposal will focus on expanding the donor numbers in this critical developmental windo w. We will employ a combination of spatial transcriptomics and multiplex immunofluorescence mapping to characterise the alterations in small endocrine object parameter s, including proliferation and proximity to ductal structures, during early life. -

Sara Sackett & Jon Odorico
University of Wisconsin-Madison
Funded by: DHHS, PHS, NIH
Probing the normal human pancreatic and islet matrisome throughout developmentDiabetes is a major world health problem, affecting millions of Americans, costing billions of dollars in treatment, and causing major late-stage health complications and death. Despite the promise of stem cell therapies for diabetes, the field of beta cell differentiation has not yet reached clinical application, mainly due to the lack of mature function and survival of the cells in vivo. A better understanding of the pancreatic and islet extracellular matrix (ECM), and what role ECM plays during beta cell development, function and maintenance may lead to a more translatable stem cell-derived beta cell product; as well as have important application in auto-and allo-islet transplantation.
-

Guido Sebastiani & Francesco Dotta
Fondazione Umberto di Mario/ University of Siena
Expression of Beta-Cell Dedifferentiation-Associated microRNAs in Type 2 DiabetesMicroRNAs are small endogenous RNAs which regulate gene expression through mRNA decay or translational inhibition. It has been previously demonstrated that microRNAs are pivotal regulators of cell phenotype and fate and are involved in many biological processes such as proliferation, differentiation or apoptosis in a wide type of cells and organisms. Given their prominent role in cell phenotype specification, our hypothesis is that microRNAs may contribute to the new recently described mechanism of beta-cell dysfunction, known as dedifferentiation, in type 2 diabetes (T2D). Indeed, in an in-vitro model of human pancreatic islets dedifferentiation, we demonstrated that microRNAs may be involved in the acquisition of islet cells undifferentiated phenotype, thus possibly playing a role in the dedifferentiation of human pancreatic islets in T2D also. Therefore, here we aim at characterizing microRNA signatures of T2D pancreatic islets in order to reveal whether microRNAs involved in in-vitro dedifferentiation of human pancreatic islets are also likely to be involved in the dedifferentiation of those islets deriving from T2D donors. In order to do this, we will specifically capture islets from pancreatic frozen sections of non-diabetic and T2D donors using Laser Capture Microdissection (LCM), and analyze microRNA profiles using high throughput technologies. The major outcome of this study will be to potentially highlight several microRNAs which may be involved in the dedifferentiation process of human pancreatic islets in T2D, thus shedding light on new possible mechanisms of beta-cell dysfunction and consequently discover new potential therapeutic target.
-

Andrew Stewart, Nathalie Fiaschi-Taesch
Icahn School of Medicine at Mount Sinai
Funded by: Breakthrough T1D and NIDDK
Multidisciplinary approaches to driving human beta cell replicationUnderstanding and controlling human beta cell replication and regeneration are major goals of the Breakthrough T1D specifically, and of the diabetes community in general. Whereas the field of beta cell replication is a relatively new one, and much new data has recently accumulated regarding the molecular control of rodent beta cells, very little is known regarding the molecular mechanisms that govern human beta cell replication. We have received support from the Breakthrough T1D and the NIDDK to specifically define the presence and subcellular location of all human cell cycle control molecules in the human beta cell. The major cell cycle control point in all cells is the “G1/S checkpoint”, or the “pRb pathway”, which contains the cell cycle proteins pRb, p107, p130, p15, p16, p18, p19, p21, p27, p57, p53, cyclins A,D,E, and cdks 1,2,4,6. Until recently, very little has been done to elucidate the presence, absence or role of these molecules in physiologic human beta cell regeneration, nor in attempts at therapeutic human beta cell replication. We are therefore trying to develop a “Human G1/S Proteome” Atlas, a task we have largely completed using human cadaveric islets provided by the Breakthrough T1D and by the IIDP. We have demonstrated that when we overexpress these cell cycle molecules using adenovirus, we can induce both robust human beta cell replication, as well as nuclear translocation of these molecules. Thus, this is an observation of great significance to beta cell regeneration investigators: it suggests that the proximate reason that human adult beta cells do not replicate and cannot be induced to replicate as that the requisite nuclear machinery does not, and apparently cannot, access the nucleus where it would drive beta cell replication. These are paradigm-changing observations, with major implications for therapeutic human beta cell replication and regeneration.\
ADDENDUM: Type 1 Diabetes results from the loss of the insulin producing beta cells. It is now clear that beta cell replacement can reverse diabetes in humans. One way to replace beta cells is to induce the regeneration of the endogenous beta cells or to expend beta cells from human donors for transplant. However, we know little about human beta cell replication. Therefore, information on human beta cell replication is critical for the development of new therapies for diabetes. Adult human beta cells proliferate at an extremely slow pace and are mysteriously resistant to the induction of replication. Rare examples of physiological replication of human cells are during the first years after birth and during pregnancy. Recently, we have made the novel and unexpected observation that many key cell cycle accelerator proteins are excluded from the nucleus in adult human pancreatic beta cells. It is possible to induce some replication of adult human beta cells by manipulating the cell cycle. However, this induction of replication is dramatically reduced during diabetes, such as Type 2 Diabetes. We propose to define the expression of cell cycle molecules in neonatal human beta cells, or human beta cells from pregnant donors or Type 2 diabetic donors. The overall goal of our research is to identify therapeutic targets to promote human pancreatic beta cell proliferation.
-

Tamás Röszer
Ulm University
Distribution of neuropeptide FF in the human infant pancreasImproving adipose tissue (AT) insulin resistance and induce adipocyte browning are key therapeutic aims in the management of obesity associated insulin resistance (IR) and in type 2 diabetes (T2DM). Our laboratory has recently identified neuropeptide FF (NPFF) as a pancreatic hormone, which selectively improves IR in the AT and controls AT browning, especially in infancy (J Clin Invest 2017, 127(7):2842-2854). The selective effect on adipose tissue is due to the specific and high level of expression of an NPFF receptor (NPFFR2) by AT macrophages (ATMs). NPFF/NPFFR2 signaling leads to a metabolically healthy ATM quaility in the AT. We have shown that differentiation of AT in infancy is controlled by ATMs, and this protects the infant from obesity (J Clin Invest 2019, 129(6):2485-2499). We also have found that NPFF controls ATM function in infancy. Next, we aim to explore whether NPFF can protect from childhood obesity (J Clin Invest 2019, 129(6):2485-2499). We already have shown the presence of NPFF in adult human pancreatic islets (J Clin Invest 2017, 127(7):2842-2854) and in adult and infant mouse pancreatic islets. Now, we would like to test whether NPFF is expressed in the infant human pancreatic islets. This is important since our data suggest that the endocrine pancreas is the major source of NPFF in the adult human plasma.
-

Yue Wang
Florida State University
Harnessing the power of beta cell regeneration to treat diabetesApproximately 1 in 11 people worldwide is afflicted by diabetes, a disease caused by a shortage of beta cells. The current standard of care for patients with severe diabetes is insulin therapy or pancreatic islet transplantation. However, insulin therapy requires frequent insulin injection, detailed dose calibration and close monitoring of blood glucose; for islet transplantation, there is a global shortage of organ donors and the procedure requires lifelong immunosuppression. As an alternative way to treat diabetes, beta cell regeneration methods have been explored with the aim to compensate for the underlying beta cell deficiency of the disease.
Human beta cells possess the potential to regenerate during normal cell turnover and under various physiological stresses. Therefore, to promote beta cell proliferation, one of the powerful approaches is to exploit the endogenous machinery of beta cell replication. However, research on human beta cell regeneration is hampered by the facts that in adult human, the basal rate of beta cell replication is less than 0.1% and bulk assays do not have enough sensitivity to distinguish signals from these rare proliferation events. To address this problem, we will develop and implement an experimental system to achieve targeted isolation of proliferating beta cells from primary human pancreas and characterize their global gene expression program.
Our work will help to identify novel pathways to increase beta cell mass both in the islet transplantation context and directly in patients with diabetes who have residual beta cells.
Core Lab
-

Irina Kusmartseva
University of Florida
Funded by: Breakthrough T1D, NIH
Pancreatic immunologic and metabolic parametersThe Organ Processing and Pathology Core (OPPC), located at the University of Florida, receives nPOD donor tissue directly from OPOs, processes the tissue, ships samples directly to investigators and stores case samples and data.
-

Daniela Del Gaudio
University of Chicago
MODY mutation testingPerform Maturity Onset Diabetes of the Young (MODY) testing for select nPOD samples.
-

Ben Giepmans
University Medical Center Groningen
Funded by: Breakthrough T1D and STW
Nanotomy of human islets of Langerhans in type 1 diabetesMicroscopy is of paramount importance in diabetes research: Paul Langerhans was the first to describe (1869) Islets based on a light microscopic (LM) evaluation of the pancreas. (Immuno-)EM-analysis of the Islets of Langerhans is an outstanding way to discriminate beta-cells from other endocrine cells: The crystalline appearance of insulin granules makes beta-cells readily distinguishable from other cells, allowing to assess total insulin content. In addition, analysis of environmental factors hypothesized to caused diabetes, e.g. viral infection, can be directly visualised. Ultrastructural analysis by EM reveals beta-cell condition, ER stress, abnormalities in mitochondria, fibrosis, as well as apoptosis, necrosis, macro- and micro-autophagy. EM can be used to follow the etiology of T1DM, the fate of transplanted Islets in both human as well as animal models and to assess the success of cellular trans differentiation to create insulin-producing cells. Moreover, EM allows other cell-types to be recognized in the Islet that affect the functioning of beta-cells, including immune cells attacking beta-cells, duct cells, endothelial cells and nerve cells. Our proposed approach enables identification of morphological and macromolecular characteristics in different cell-types within the Islets in a quantitative and high-throughput manner. A single dataset could resolve most of the features raised above. So why is EM-analysis not standard included? Traditionally, EM data-interpretation is mainly done by the publishing scientist(s). EM-examination is hampered by the laborious procedure that requires technical expertise and expensive equipment. In addition, data-sharing is limited, the field of view of the area inspected at high magnification is traditionally very small, the data is noisy making it difficult to objectively quantify and segment the greyscale EM-images. Without the knowledge of how Islets of Langerhans look like, it will remain cumbersome to establish the cause and solution to diabetes. Implementation of computational science has now revolutionized EM, paving the path for routine EM-analysis of complete Islets, as we have pioneered.
We are creating and analyzing high-resolution EM-maps of cross-sections of Islets of Langerhans in health and diabetes, which we term Nanotomy of Islets. The datasets allow to determine many aspects, including leukocyte infiltration, insulin content and cellular stress. Importantly, small particles such as viruses will also be detectable. The data first will be shared with nPOD co-investigators and at a later stage will be made publically available. (adopted by nPOD staff)
-

Clive Wasserfall
University of Florida
UF AAb CoreThe Autoantibody Core at the University of Florida serves as the coordinating center for nPOD screening lab partners (LABS,Inc and Viracor-IBT Laboratories). Combi-kit manufacturing and quality control/assurance measures are managed here. Additionally, the UF Autoantibody Core tests all nPOD samples for autoantibodies to GAD, IA-2, and ZnT8 by ELISA.
-

William E. Winter
University of Florida
Northwest Lipid ResearchThe Marcovina lab performs C-peptide testing for all nPOD samples.
-

Liping Yu
University of Colorado-Denver
Denver AAb CoreThe Autoantibody Core at the University of Colorado works with the Autoantibody Core at UF for quality control/assurance and tests all nPOD samples for autoantibodies to GAD, IA-2, Insulin, and ZnT8, by RIA.
Exocrine Pancreas
-

Brittany Bruggeman & Martha Campbell-Thompson
University of Florida
Congruence of lab versus histopathologic findings of acute pancreatitis in the nPOD consortiumAcute pancreatitis is the cause of more than 200,000 hospital admissions in the United States each year (1). Acute pancreatitis presents very broadly, hence diagnosis can be difficult and requires confirmatory tests. Serum amylase and lipase levels have been established as markers for disease. However, they lack adequate specificity and sensitivity which might make their use insufficient in providing the most accurate diagnosis.
This study aims to investigate whether varying degrees of elevation in lipase and amylase levels can serve as more accurate predictors of acute pancreatitis by histopathology. Additionally, the study seeks to identify other factors that can predict elevated amylase and lipase levels during the hospital course of deceased organ donors, as well as explore the clinical characteristics of donors who have acute pancreatitis based on histopathological examination.
This project will consist of an nPOD chart review to evaluate amylase and lipase levels in critically ill patients as predictors of acute pancreatitis by histology, and other factors that may contribute to elevated amylase/lipase levels such as DKA, renal failure, and GI illness. Our main hypothesis is that if the lipase levels in a person’s blood during their hospital stay are three times higher than the upper normal limit, it could be a good indicator of acute pancreatitis by histopathology. However, if the amylase levels are generally high or if the lipase levels are less than three times the upper normal limit, it may not accurately predict the presence of acute pancreatitis.
-

Anna Casu
AdventHealth
Funded by: Breakthrough T1D
Stellate cells in type 1 diabetes research (STELLAR)The loss of insulin-producing β-cells is central to the pathogenesis of type 1 diabetes (T1D). More recently, the contribution of the exocrine pancreatic cellular components is becoming evident in determining the β-cells well-being. Other pancreatic cell types could be acting by reducing β-cell resistance to the autoimmune attack or inducing a stressful condition that will lead to neo-antigen expression or modulate the autoimmune response favoring β-cell damage.
The pancreatic stellate cells (PSCs) reside in the exocrine pancreas in the peri-acinar space, but also in perivascular and periductal locations. They are implicated in the pathobiology of major exocrine pancreatic disorders such as chronic pancreatitis and pancreatic cancer. Activated PSCs lose their vitamin A content and are responsible for organ fibrosis, promote extracellular matrix deposition, release factors that promote an inflammatory state, interfere with acinar cell function, and reduce the insulin secretory capacity. The PSC activity known in other diseases leads to hypothesize that they could have a role either in determining the exocrine pancreas alterations seen in T1D and favoring β-cell function decline. In spite of the multiple possible causative mechanisms, the role of PSCs in the pathogenesis of T1D has not been investigated, and this proposal aims to close this knowledge gap.
A preliminary analysis of a subset of HPAP single-cell data showed that PSCs from organ donors with T1D have an immune-activated phenotype as described by Baron et al. in other conditions. They overexpress in fact multiple chemokines and IL-6. This evidence supports our hypothesis that a dysfunctional and proinflammatory PSC activation in T1D contributes to and maintains β-cell stress accelerating β-cell functional decline. It is hypothesized that this interaction is mediated by PSC-secreted factors directly acting on the islets which promote a pro-inflammatory gene expression. In this study, we will investigate the role of activated PSC-secreted factors on islet function.
-

Hsun Teresa Ku
City of Hope
Funded by: NIH/NIDDK
The role of human ductal cells in propagating pathogenesis of type 1 diabetesType 1 diabetes (T1D) is a pancreatic disease marked by the autoimmune destruction of insulin – secreting beta cells within the endocrine compartment, leading to chronic hyperglycemia and long – term complications. Despite extensive studies into the autoimmuni ty that target s beta cells, a complete understanding of the pathogenesis of T1D remains unclear . A recent study by Fasolino et al. demonstrated that the exocrine ductal cells in T1D donors , compared to non – diseased control donor s, have an elevated expression of a MHC class II molecule, HLA – DR. MHC class II molecules are known to be expressed by antigen presenting cells (APCs) that process and present extracellular antigens to CD4 – expressing T helper cells. Fasolino et al. also found that CD4+ T cells , compared to CD8+ T effector cells, are located more frequently in the neighborhood of HLA – DR expressing ductal cells . These results raised the intriguing possibility that pancreatic ductal cells may function as APCs. In this study, we propose to conduct a more comprehensive assessment of proteins involved in antigen processing and presentation in pancreatic ductal cells of T1D donor t issues , compared to control non – diseased donor tissue s , using co – immunofluorescence staining technique . Our proposed studies, if successful, will contribute to a new line of research addressing the cross talk between exocrine and endocrine pancreas in the pathogenesis of T1D , which may lead to new therapeutic target s for T1D. -

Kathrin Maedler
University of Bremen
Funded by: DFG
Do enteroviruses trigger diabetes progression?Identification of the driver of autoimmunity and early intervention before established disease is urgently needed. As in many autoimmune diseases, viral infections have been associated with T1D, recently confirmed by large and highly sensitive screenings u sing well – characterized material from autopsy and biopsy. W e looked closer into the pancreases from organ donors with early T1D – associated autoimmunity before T1D diagnosis and with established T1D and found the factor YAP, co – transcriptional activator of the Hippo cell and organ developmental pathway highly incr eased. Absent in mature functional beta – cells, YAP was especially found in areas of enterovirus – infected cells which remain deposited in the pancreas long after acute viral infection and drive inflammation and immune cell infiltration. Such changes in the exocrine pancreas have been completely overlooked. To our surprise, in a scenario of T1D, YAP did not balance the immune response but acted oppositely of what has previously been known; it potentiated the deleterious effect of the enterovirus and constituted a vicious cycle; YAP increased virus load, infla mmation and cell death in both exocrine and endocrine cells. Eventually, even YAP + – cells migrated into islets. Our project shifts two paradigms: we hypothesize that (1) autoimmune diabetes starts in the exocrine pancreas with areas of inflammation and immune infiltration and (2) YAP acts as early driver of autoimmunity and T1D by potentiating inflammatory signals t owards immune cell attack and damage to beta – cells. Our analyses will explain the early pathology seen very early even in first degree relatives (FDR) of patients with T1D, who have already reduced pancreas mass. The objective of this exploratory project is to unravel how YAP expression in viral+ cells in the pancreas fosters T1D initiation and progression and how this cycle of inflammation can be interrupted to establish an early pharmaceutical intervention against autoimmunity and T1D.
Immunology
-

Reza Abdi & JianJun Cheng
Brigham and Wowen's hospital
Use of T1D pancreastic and lymphoid tissue to assess the expression of adhesion moleculesWhile immunoregulatory therapies hold great promise for the treatment of T1D, their inadequacy and serious toxicity have limited research efforts to find a lifelong immunosuppression approach. This has motivated investigators to search for alternative approaches such as the use of nanotechnology to deliver a wide variety of drugs and biomolecules. In recent years, there have been major strides to address various obstacles in order to bring nanotechnology closer to clinical practice. Nanotechnology has been moving forward on two fronts: 1) synthesis of a NP which efficiently loads, retains and releases drugs and 2) development of an innovative strategy to deliver NP to specific sites. Much progress has been made on NP surface properties and size to reduce burst release, blood opsonization and clearance to enhance the circulation half-life of NP from several minutes to tens of hours.
On the front of delivering NPs to specific tissue sites, we have witnessed less progress. To make our nano-delivery approach applicable to Type 1 diabetes (T1D), we (Abdi and Cheng) have made major strides as follows: (A) we have successfully designed nanomedicines with the desired surface properties and controlled particle sizes to improve their pharmacokinetics, (B) demonstrated that loading and controlled release of the drug is markedly improved by developing a polylactide-drug conjugated nanoparticle, or nanoconjugate, via drug-initiated polymerization of lactide followed by nanoprecipitation, (C) our published data indicate that our nanotechnology platform is highly efficient in delivering immunosuppressive drugs, and importantly..
(D) Importantly, our preliminary data indicate selective PNAd dependent delivery of the nanoparticles to the pancreas and pancreatic lymph nodes in NOD mice. We are interested in examining the expression of PNAd in pancreatic lymph nodes (PLN), pancreas, non draining LN and spleen by immunhistolgoy to assess the feasibility and potential success of our targeted therapy in humans.
-

Medhi Benkhala & Alberto Pugliese
City of Hope
Funded by: City of Hope
Enterovirus infection and pancreatic macrophage phenotypes in type 1 diabetesType 1 diabetes (T1D) is a chronic autoimmune disease targeting pancreatic beta cells. The incidence of T1D has been increasing, a trend inexplicable by genetic predisposition alone, indicating that environmental factors such as viruses play important roles in triggering the disease. Decades of research have provided epidemiological evidence for an association between T1D and enteroviruses, particularly coxsackieviruses. As shown in nPOD donors , T1D is associated with higher rates of viral RNA, capsid protein, and associated immune markers compared to non – diabetic cont rols. However, there remains limited mechanistic evidence establishing how viral infections disrupt immune homeostasis to enable autoimmune destruction. Recent evidence demonstrates that pancreatic immune homeostasis depends on regulatory interactions betw een tissue – resident memory T cells and macrophages. W e hypothesize that enterovirus infections convert protective tissue – resident macrophages to pathogenic phenotypes, disrupting regulatory networks that normally prevent autoimmune T cell activation agains t beta cells. We will examine archived pancreatic tissue sections from nPOD. We will compar e non – diabetic donors and donors with T1D, specifically examining donors with or without detectable enterovirus infection (VP1+ vs VP1 – ) as identified by the nPOD – virus group. We will also exploit the organ donor pancreas slice platform, which is well established in our laboratory, as this is ideally suited to test this hypothesis in human pancreas tissue. Pancreatic tissue slices are thick sections of pancreas that e nable functional analysis of live tissue in which the immune component in the pancreas is preserved and can be investigated; this includes tissue – resident macrophages and T cells. The study of pancreas slices enables the ability to investigate both functional and spatial interactions as the tissue architecture is preserved . Overall, this investigation will generate mechanistic insights into how enterovirus infection disrupts tissue – resident macrophage regulatory networks in human pancreatic tissue, establis hing critical preliminary data linking viral triggers to the initiation of autoimmune diabetes. These findings will inform future comprehensive studies of therapeutic interventions targeting macrophage phenotypes and immune checkpoint pathways to preserve protective immune networks and prevent irreversible beta cell loss. -

Todd Brusko
University of Florida
Funded by: University of Florida
Molecular Signature of Autoimmune T cells in Type 1 Diabetes and Treg TCR deep sequencing. Detect Cd226+ Treg and Teff ininsulitis patient samplesThe Brusko Lab is generally interested in the cellular immune response involved in the disease process leading to type 1 diabetes (T1D). A strong genetic association between T1D and the MHC class II gene region of the adaptive immune system has implicated coordinated T and B cell responses as a central mediator of the disease process. To date, the majority of studies in humans have been limited to investigations using peripheral blood samples. While these studies have identified a number of T cells reactive to autoantigen targets (e.g., insulin, glutamic acid decarboxylase (GAD), islet antigen-2 (IA-2), zinc transporter-8 (ZnT8), among others), they have been unable to identify clonal signatures unique to T1D patients. In an effort to generate a disease specific signature, our lab initiated studies to isolate CD19+ B cells, CD8+ T cells, CD4+ T conventional cells (Tconv), and CD4+CD25+CD127-/lo Tregs from nPOD-donor tissues including spleen, pancreatic draining lymph nodes, irrelevant draining lymph nodes, and peripheral blood. From this donor material, we will generate a molecular profile of T1D including a characterization of the T cell receptor (TCR) and B cell receptor (BCR) repertoire, global DNA methylation profile, and transcriptional signature. We believe that this effort will lead to novel insights into the specificity and functional properties of immune cells that lead to autoreactive beta cell destruction. Of particular interest, we have been focused on a T cell costimulatory pathway involving CD226 or TIGIT interacting with CD155. We believe the molecular signatures identified in cell subsets isolated from the site of autoimmune attack (e.g., the islets and draining lymph nodes) will provide insight into the cause of immunoregulatory defects leading to T1D.
-
Todd Brusko
University of Florida
Funded by: Helmsley Charitable Trust
Antigen discovery for regulatory T cells in T1DThe Brusko Lab is generally interested in the cellular immune response involved in the disease process leading to type 1 diabetes (T1D). A strong genetic association between T1D and the MHC class II gene region of the adaptive immune system has implicated coordinated T and B cell responses as a central mediator of the disease process. To date, the majority of studies in humans have been limited to investigations using peripheral blood samples. While these studies have identified a number of T cells reactive to autoantigen targets (e.g., insulin, glutamic acid decarboxylase (GAD), islet antigen-2 (IA-2), zinc transporter-8 (ZnT8), among others), they have been unable to identify clonal signatures unique to T1D patients. In an effort to generate a disease specific signature, our lab initiated studies to isolate CD19+ B cells, CD8+ T cells, CD4+ T conventional cells (Tconv), and CD4+CD25+CD127-/lo Tregs from nPOD-donor tissues including spleen, pancreatic draining lymph nodes, irrelevant draining lymph nodes, and peripheral blood. We are also interested in using single cell ‘omics to associate phenotypes with immune receptor profiles in draining lymph node, pancreas, and pancreatic islets. From this donor material, we will generate a molecular profile of T1D including a characterization of the T cell receptor (TCR) and B cell receptor (BCR) repertoire, global DNA methylation profile, and transcriptional signature. We believe that this effort will lead to novel insights into the specificity and functional properties of immune cells that lead to autoreactive beta cell destruction. Of particular interest, we have been focused on a T cell costimulatory pathway involving CD226 or TIGIT interacting with CD155. We believe the molecular signatures identified in cell subsets isolated from the site of autoimmune attack (e.g., the islets and draining lymph nodes) will provide insight into the cause of immunoregulatory defects leading to T1D.
-

Georgia Fousteri
San Raffaele Research Institute
Funded by: Ministero della Salute, Giovani Ricercatori
T follicular helper (TFH) and regulatory (TFR) cells in T1DType 1 Diabetes (T1D) is characterized by the gradual loss of insulin-producing beta cells, which are eliminated by autoreactive cells infiltrating the pancreas. In humans and mouse models of the disease, T1D susceptibility is determined by genetic factors and is greatly influenced by environmental triggers (1). Progression to T1D is usually preceded by a period of anti-islet autoantibody production, which often can last for years (2). Today, autoantibodies are most widely used as serum biomarker (3), but T-cell readouts and metabolome studies are underway as they may strengthen diagnosis, prognosis and most importantly bring forward effective protocols for prevention and cure. T follicular helper (TFH) cells are essential for the formation of germinal centers (GCs), specialized structures in secondary lymphoid organs where maturation of B cells into high-affinity plasma cells and long-lived memory B cells takes place. It has become clear that precise control of the GC response is important for the production of optimal antibody responses that are devoid of selfreactivity. The exact mechanism by which autoreactive B cells are eliminated during this process is not fully understood, however the number of TFH cells together with a specialized subset to FOXP3+ regulatory T cells, namely T follicular regulatory cells (TFR), finely control the survival of pathogen-specific high-affinity B cells. It is currently not known whether TFH or TFR cells are involved in the production of islet-reactive, autoantibody-producing B cells in patients with T1D. However, given that autoantibodies emerge prior to disease onset, the effect of TFH cells in T1D progression can be inferred. With this study we aim at identifying whether TFH and TFR cells are implicated in autoantibody development in T1D. Investigating this mechanism poses a cardinal opportunity to increase the pipeline of therapeutic targets and to improve the number of disease biomarkers.
-

Rachel Friedman
University of Colorado
Funded by: Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
Mononuclear phagocyte populations in T1D isletsLittle is known about how T cell pathogenesis is modulated in the islets during T1D, yet this is a critical obstacle for preventing destruction of remaining islets or beta cell grafts. Therefore, we must elucidate mechanisms that promote and prevent the pathogenic autoimmune response in the islets. Our data in the NOD mouse model show that different populations of CD11c mononuclear phagocytes have countervailing roles in restimulating or inducing tolerance in islet-infiltrating T cells as islet infiltration progresses. While macrophages have been identified in human pancreatic islets, these likely represent a complex assortment of several mononuclear phagocytic cell populations that play a role in islet destruction. As a result, our goal is to identify the populations of mononuclear phagocytes within the islets of normal, autoantibody positive, and type 1 diabetes subjects.
ADDENDUM: Type 1 diabetes (T1D) lacks therapeutic approaches that stop the autoimmune destruction of the beta cells. T cells may be the major mediator of destruction in T1D, but they do not act alone. T cells are influenced by myeloid cells in the islets. Our preliminary data show that human islets contain multiple populations of myeloid cells with some populations that are only present in the islets of T1D patients. As a key regulator of T cells in T1D, the islet myeloid cells are a potential target for therapeutic intervention for T1D. However, human islet myeloid cells are severely understudied. Thus, before we can develop myeloid therapies for T1D, we must understand the human islet myeloid populations and identify specific molecules on the myeloid cells for therapeutic targeting. Our goals are to identify the myeloid cell populations in the islets of non-diabetic, autoantibody positive, and type 1 diabetic donors. We will determine where these populations are localized in the islets, as well as how these populations correlate with islet health and interact with T cells in the islets. These analyses will create a picture of the myeloid landscape of human islets and begin to elucidate the roles of human myeloid populations in normal and type 1 diabetic islets. Because myeloid cells are present in all islets, the knowledge gained will be relevant to understanding how these cell populations function in T1D and T2D.
-

Allison Green and Sarah Richardson
Hull York Medical School
Funded by: Steve Morgan Foundation
Investigation of the sexual dimorphic phenotype of complosome expressing medullary thymic epithelial cells, and thymic B cells in type 1 diabetesQuality control – the removal of immune cells that have the ability to attack our own tissues – is a central tenet of the immune system. In type 1 diabetes ( T1D), impaired quality control in the thymus, leads to release of beta cell – reactive immune cells (called T cells) into the blood; such cells facilitate destruction of insulin – producing beta cells in the pancreas. Little is known how thymic quality control fails. Medullary thymic epithelial cells (mTECs) are specialized cells in the thymus that are critical for expressing a range of self – proteins, including beta cell proteins e.g. insulin, to developing T cells enabling selection and destruction of T cells reactive to the host proteins. Loss, or impairment, in mTEC homeostasis or function leads to autoimmune conditions in mice and humans. Previously in NOD mice , a well – described animal model of T1D , we made a breakthrough in understanding why thymic quality control fails; abnormal structures enriched with immune cells called thymic B cells form, autoantibodies bind to mTECs and insulin – expressing mTECs die. Now we know that mTEC survival is linked to activation of intracellular complement component C3 (termed complosome) in mTECs, and there is an undefined relationship between thymic B cells, mTEC complosome activity and mTEC survival. We have initiated translation of our findings to humans; human mTECs express C3 from fetal to adult life correl ating with an increase in thymic B cells , and complosome activity is detectable in human thymic tissue sourced from non – diabetic donors. In stage 1 of this pro ject , using a multiplexing immunhistochemical approach we will determine if beta cell – specific protein expression and mTEC survival is linked to impaired C3/complosome activity in nPOD donors without diabetes at various ages and in both males and females. In Stage 2, we will expand the number of ND donors and determine if these characteristics are altered in AAb+ and T1D donors . Further , we will analyse thymic B cells (number, location and phenotype) , to determine if ‘rogue’ thymic B cells correlate with T1D development. -

Kathryn Haskins, Sally Kent & Thomas Delong
University of Colorado School of Medicine
Hybrid peptides as target antigens for pathogenic T cells in human T1D patientsWe have recently discovered a new form of beta-cell autoantigen, which is recognized by CD4 T cells, and is formed through a novel post-translational modification of beta-cell peptides that involves the fusion between insulin peptide fragments and peptides of other secretory granule proteins. The hybrid insulin peptides (HIPs) are highly antigenic for autoreactive and pathogenic CD4 T cell clones derived from the nonobese diabetic (NOD) mouse. Importantly, three of these HIPs have been shown to exist in nature, as we detected them by mass spectrometry of antigenic fractions from beta cell extracts. Moreover, we have also identified two HIPs that are antigenic for T cells clones derived from the islets of T1D deceased donors. Thus, our data support the involvement of HIPs in human T1D. In this project, our goal is to extend our study of human T cell responses to HIPs by isolating T cells from spleen and pancreatic lymph nodes (PLN) of nPOD donors and then testing these T cells on a selected panel of HIPs. We will initially test the T cells through a sensitive assay (called ELISpot) for the inflammatory cytokine, interferon-γ, produced when CD4 T cells encounter antigen. Long-term goals are to identify HIPs that stimulate autoreactive T cells from human patients and to develop reagents with these HIPs that can detect the autoreactive T cells in the blood (or other tissues).
-

Eddie James
Benaroya Research Institute
Characterization of T Cells that Recognize Post-Translationally Modified Beta Cell Epitopes from within the PLNWe have recently demonstrated that enzymatically modified peptides derived from beta cell antigens are preferentially recognized by autoreactive CD4+ T cell clones isolated from the peripheral blood of patients with T1D. Furthermore, T cells that recognize these modified peptides are present in the peripheral blood of patients with T1D and have a high affinity, enabling their detection by flow cytometry. Importantly, the expression and activity of enzymes responsible for these modifications are upregulated in response to cellular stress and inflammation. Therefore, modified peptides are likely to form in stressed islets and to elicit pathogenic T cell responses. Consistent with this notion, T cells that recognize two or three of these modified peptides were shown to be present in islets and pancreatic lymph nodes (PLN) of two organ donors with T1D. The goal of this project is to corroborate and extend our study of T cell responses to modified beta cell antigens are relevant to the pathogenesis of T1D by identifying and studying T cells that recognize these peptides within PLN from nPOD donors. We will identify and isolate these T cells using HLA class II tetramers. We will then characterize these cells by examining their cytokine profiles and sequencing their T cell receptors. Finally, we will compare these attributes with those of T cells with the same peptide specificity that are found in peripheral blood to define unique attributes of T cells that are most relevant to disease.
-

Jenny Jiang & Todd Brusko
The University of Texas at Austin
Human T cell repertoire profiling in Type 1 DiabetesCurrent strategies of studying islet autoantigen-specific T cells rely heavily on fluorescence labeled tetramers and are limited in the throughput of antigens that can be analyzed simultaneously. The profile of islet autoantigen-specific T cells that can be obtained is limited. We recently developed a new technology, Tetramer associated TCR sequencing (TetTCR-Seq), that is capable of de novo generating a large amount of peptides and mapping antigen recognition with single cell TCR sequences by DNA barcoded tetramers. It enables us to quickly survey T cell repertoire at single cell level for a large number of peptides. Meanwhile, simultaneous profiling of gene/protein expression at single cell level helps to provide a comprehensive evaluation of antigen-specific T cell repertoire. We propose to apply this novel technology to study islet autoantigen-specific T cells in T1D. By investigating islet autoantigen-specific T cells in different tissues from nPOD, including spleen, pancreatic draining lymph node, and irrelevant draining lymph nodes, we will characterize the antigen-specific TCR repertoire and disease-specific transcriptional signatures. We believe this will help us to gain deeper understandings of the onset of T1D, the cause of immune dysregulation and potentially develop new markers for the diagnosis and the treatment of T1D.
-

John Kearney
University of Alabama at Birmingham
Funded by: Breakthrough T1D
Modulation of autoimmune diabetes by natural antibodies specific for N-acetyl-D- glucosamineThe focus of our research project is to improve the understanding of how antigenic stimulation of the B lymphocyte repertoire development during early life may protect from Type 1 Diabetes (T1D). Specifically, we are investigating how exposure to the common pathogen Group A Streptococcus promotes the formation of a T1D-suppressive natural antibody repertoire. Exposure to microorganism associated antigens during early life can protect diabetes-prone mice from developing T1D, and epidemiological studies in humans as well as studies in diabetes prone mice and rats have reported that that immune responses to Group A Streptococcus (GAS) are associated with protection from T1D. In humans and mice, infection or immunization with GAS results in a robust B cell response and the production of antibodies reactive with GAS cell wall-polysaccharide-associated N-acetyl-D- glucosamine (GlcNAc) epitopes, yet the role of these responses in protection from T1D has not been previously examined. We have shown that GlcNAc-reactive antibodies, produced following exposure to GAS cell wall antigens, bind GlcNAc-modified beta cell antigens. GlcNAc-reactive B cells elicited during neonatal immune responses to GAS traffic to the pancreas during T1D in non-obese diabetic (NOD) mice, and transfer protection upon adoptive transfer into naïve recipients. GlcNAc-reactive natural antibodies produced by these cells opsonize damage-associated molecular patterns on stressed beta cells and promotes clearance of their diabetogenic antigens, thereby attenuating the pathogenesis of T1D. Our goal is to evaluate the role of GlcNAc-reactive B lymphocyte development and antibody production in T1D in humans to identify novel biomarkers for improved prediction of T1D susceptibility, and the development of vaccination or probiotic strategies aimed at boosting natural antibody-mediated suppression of T1D progression.
-

Sally Kent
University of Massachusetts Medical Center
Funded by: Breakthrough T1D
Investigation of B cells in human islets and PLN in T1Our goal is to understand the role of autoreactive B cells in tissues draining the pancreas and their role in the type 1 diabetes (T1D) autoimmune response. While B cells are responsible for secreting autoantibodies, B cells have other functions including secretion of effector cytokines, antigen presentation and costimulation of T cell responses. Using a multiplexed, single cell interrogation assay called microengraving, we saw, ex vivo, that there is a statistically more frequent number of immature (IgM secreting) CD20 B cells secreting antibodies reactive with islet antigen GAD65 in the spleen from 50% individuals examined with recent onset (0-7 years) T1D as compared to those from healthy control: these cells could also be seen on the spleen of some GADA+ individuals without disease and in some individuals with long-term T1D. We saw this reservoir of GADA-secreting B cells of a mature phenotype (secreting IgG1, IgG3 or IgA) in the spleen of two individuals with recent onset T1D. We saw a low frequency of immature CD20 B cells secreting antibodies reactive with proinsulin in all groups. We wished to interrogate the autoreactive B cells for their cytokine secretion, but due to cell demise in the single cell assay at time points greater than 24 hours, we developed a 7-day culture assay that allows for survival of B cells and identification of these cells by their autoantibody secretion and cytokine profile in a two-color ELISpot assay. We are interrogating spleen samples from control individuals, individuals with autoantibody, but without disease, individuals with recent onset T1D (0-7 years), and individuals with long-term T1D (>7 years) for co-secretion of GAD65 autoantibody and IL-6/IL-10 or LTa/TNFa. These studies will provide a profile of the function of autoreactive B cells from individuals with T1D and their role in the autoreactive disease process.
-
Sally Kent
University of Massachusetts Chan Medical School
Funded by: NIDDK
T cell autoreactivity, phenotype and function directly from islets of donors with recent onset type 1 diabetes (T1D) and from donors with islet-associated autoantibodies, but without clinical diseaseThe goal of this project is to further our understanding of the CD4 T cells and CD8 T cells that infiltrate the islets in autoantibody+ donors at risk for development of type 1 diabetes (T1D) and from donors with recent onset T1D. We will hand pick islets from digests of live pancreas slices from the two cohorts of donors, supplied by nPOD and examine them in two ways. First, we will culture single islets in conditions that favor T cell outgrowth from islets: this will add to the unique bank of islet-derived T cell lines we have generated. These T cell lines can be propagated in culture and used to test reactivities against specific epitopes, both conventional and neoepitopes, for both CD4 and CD8 T cells; these tests will also reveal the islet-infiltrating T cells HLA restriction and effector phenotype. Second, hand-picked islets will be analyzed for their transcriptome by sc-RNA-Seq with TCR sequencing; this method captures more in depth phenotypic and functional information about islet-infiltrating T cell. The TCR sequencing will allow for the construction of TCR transductants which will be used to examine reactivity to conventional and neoepitope and for HLA restriction. In addition to furthering our knowledge concerning the T cells that infiltrate the site of pathology, the islets, early in the disease process, information generated here could be used in constructing antigen-specific Tregs or used in tolerance induction protocols.
-
Sally Kent & Eddie James
University of Massachusetts & Benaroya Research Institute
Funded by: Cystic Fibrosis Foundation
T cell autoreactivity in pancreatic draining lymph nodes (pLN) of donors with cystic fibrosis-related diabetes (CFRD) and type 1 diabetes (T1D)Many individuals with cystic fibrosis (CF) develop diabetes or CF – related diabetes (CFRD). Some published data indicates that CFRD shares autoimmune phenotypes in common with type 1 diabetes (T1D) . These includ e a higher prevalence of islet autoantibodies for CFRD patients than the general population and signs of broad immune dysregulation (including a Th17 signature). N otably, in autoantibody – positive CF patients, diabetes onset occurs earlier and is generally associated with a higher risk of acute complications . In a study funded by the Cystic Fibrosis Foundation, T cells from peripheral blood mononuclear cells from matched cohorts of individuals with CFRD, with CF, with T1D, and from controls are being examined for islet – reactivity and phenotype. Here, we propose to examine cryopreserved single cell suspensions of pancreatic draining lymph nodes from matched donors with CFRD, with T1D, and from donors without diabetes with and without pancreatitis (the latter serving as an inflammation control) for these paprameters. Our overall goal is to increase the understanding of the autoimmune response to islet – antigens in donors with CFRD and in T1D as compared to those from control and to those from donors with pancreatitis (inflammatory state) and from control donors and identify shared T cell specificities and immune mechanisms that might contribute to their decreased insulin secretion and beta cell damage i n CFRD and T1D . -
Pia Leete & Joanna Boldison
University of Exeter Medical School
Funded by: RD Lawrence Fellowship Diabetes UK & Leona M. and Hary B. Helmsley Charitable Trust
The Heterogeneity of Type 1 Diabetes: An Islets StoryWhen a person is diagnosed with Type 1 diabetes (T1D), t he amount of beta – cell mass each individual lose s is highly variable despite their clinical need for insulin therapy. Attempts to detect robust markers in the blood that precisely describe disease activity in the islets are ongoing , but without a full understanding of the disease course in the pancreas, when attempting to target therapy to the organ , we are still operating par tially blindfolded. We have previously reported that two forms, or endotypes, of T1D are evident in the pancreas [1 – 4] . T hese differ according to the degree of beta – cell loss, the composition of the islet – specific immune infiltrate , and how beta cells process insulin . Additionally, we now have preliminary data that suggests the beta – c ells them se lves may vary between the two forms ) , even hint ing that successful beta – cell regeneration may be more likely in one form than the other . Our proposed study will expand on these investigations , and we will compare pancreatic protein expression profiles between people diagnosed with T1D at different ages, with different durations of disease and compare this to those without diabetes and with Type 2 diabetes (as a proxy for metabolic dysregulation) . We will leverage advanced histological and imaging techniques to analyse the presence of multiple critical proteins in granular detail in whole sections of pancreas . W e will then (b ased on several features of pancreatic T1D and beta – cell loss ) , construct a nuanced ‘pseudo’ timeline of disease progression in T1D to illustrate the life and loss of beta – cells in T1D and explo re if this is the same in all p eople, randomly different between people – or st ra tifiably different between gro ups of people living with T1D . With this information we will bett er understa nd if we should be looking for distin ct forms of T1D , and this may h elp scientists and trialist s underst and why some people re spond brilliantly in clini cal trials, whilst other do not. -

Enrique Montero
Arthur Riggs Diabetes & Metabolism Research Institute
CD6 and its ligands in Type 1 DiabetesType 1 diabetes mellitus (T1D) results from the autoimmune attack of pancreatic β-cells, resulting in a loss of functional β-cell mass. The CD6 molecule is a membrane glycoprotein predominantly expressed on T cells and is implicated in lymphocyte adhesion, activation, and proinflammatory commitment. The CD6 ligand CD166/ALCAM is upregulated in mature dendritic cells and the inflamed tissue, facilitating infiltrating lymphocytes into the target organs. Furthermore, CD166 is implicated as essential in pancreatic development, and it has been found in the membrane of islet endocrine cells.
CD318/CDCP1 was recently identified as a second CD6 ligand, contributing to the autoimmune response in experimental models. We have discovered in healthy human donors that CD318 is expressed by a subset of myeloid Dendritic Cells and activated monocytes. Moreover, CD318 expression can be induced on peripheral monocytes by proinflammatory cytokines. CD318+ cells significantly reduced the proliferation of polyclonal T cells and antigen-specific CD4+ T cells. Our unpublished data also uncover CD318 expression in human pancreatic islets from non-diabetic donors, and its expression is increased by proinflammatory cytokines.
These findings suggest a novel role for CD318 as a co-inhibitory ligand for T cell activation and effector function. Therefore, we propose CD6 as a novel immune checkpoint with dual function based on the ligand expression: CD6 binding to CD166 triggers T cell inflammatory response in the pancreas while CD318 acts as a co-inhibitory ligand down-regulating T cell responses and preventing the perpetuation of local autoimmunity. This project aims to characterize the expression of CD6 and its ligands CD318 and CD166 in human pancreatic islets from T1D donors. This study may describe a novel natural mechanism for human islet protection from autoimmunity and may aid developing novel therapeutic strategies to cure T1D by targeting CD6.
-

Maki Nakayama
University of Colorado
Dynamic Survey of T Cell Repertoire Targeting Pancreatic Beta CellsOur project aims to define islet-specific T cell receptor (TCR) sequence repertoires directly from pancreas to pursue two goals; 1) to identify antigens targeted by T cells contributing to the development of type 1 diabetes (T1D); and 2) to pursue the potential of TCR sequences to be used as biomarker. We hypothesize that common antigens targeted by T cells negatively (Treg) and positively (Tpath) contributing to the development of T1D in multiple patients are present. Identification of such antigens and TCR repertoires will lead us to develop antigen-specific immunotherapy and immunodiagnostic tools. We directly sequence TCR alpha and beta chains of T cells in the pancreas as well as in different T cell subsets in the immune organs and peripheral blood of patients having T1D utilizing the high-throughput Illumina and 454 sequencing technologies. TCRs detected in the pancreas are expressed on TCR-null T cell hybridoma cells using the retroviral expression system and are tested for the response to known antigens as well as the islet cells. Once TCR pairs responding to the islet cells are determined, we will further assess pathogenicity in inducing diabetes by generating retrogenic animals. In addition, presence and frequency of pancreas-derived TCR sequences in the peripheral organs of the same patients as well as those having the same HLA alleles will be evaluated to “trace” T cells detected in the pancreas. Given evidence that a large number of public TCR sequences detected from multiple individuals are present, we believe that TCRs associated with the development of T1D exist and are able to be used for diagnosis in combination with other T cell assays (e.g. tetramer analysis). Common antigens associated with the pathogenesis and regulation of T1D development will gain our insights to develop antigen-specific immunotherapy for T1D.
-
Maki Nakayama & Mia Smith
University of Colorado Anschutz Medical Campus
Funded by: NIDDK
Immune receptor repertoires associated with type 1 diabetes progressionImmune receptors, including T cell receptors (TCRs) on T cells and B cell receptors (BCRs) on B cells, are central to immune responses, mediating antigen recognition and tissue specificity. Characterizing the immune receptor repertoires during type 1 diabetes (T1D) progression provides key insights into the T and B cell populations contributing to disease pathogenesis. The overarching goal of this project is to identify and characterize the immune receptors expressed by T and B cells infiltrating the pancreas and islets at various stages of T1D.
Over the past decade, we have identified TCR sequences in the pancreas of stage 3 T1D donors (those requiring insulin therapy) and non-diabetic individuals. We found that only diabetic donors possess T cells recognizing both native and modified forms of (pre-pro)insulin, highlighting insulin and its derivertives as key targets in beta cell autoimmunity. These results underscore the crucial role of insulin and its processing forms as autoimmune targets in T1D. However, due to the low frequency of islet-specific T cells in the pancreas and the significant antigenic heterogeneity observed between individuals, further investigation is necessary to delineate the immune receptors involved in T1D.
Two critical questions remain: (1) What are the immune receptors and their target antigens expressed by T and B cells during the earlier stages of T1D? and (2) What are the phenotypic characteristics of T and B cells expressing islet-specific immune receptors?
To address these questions, we propose to:
- Identify immune receptors expressed by T and B cells in the pancreas of donors at T1D different stages and investigate their antigen specificity.
- Profile gene expression associated with immune receptor signatures in individual T and B cells from pancreatic lymph nodes.
Completion of this study will provide crucial insights into the antigen specificity required for the adaptive immune system in T1D development, and inform strategies for immunotherapy and biomarker development for T1D.
-

Mark Peakman
Sanofi
Credentialing immunotherapy targets in islet tissues with insulitisSanofi is developing strategies to control beta cell destruction in type 1 diabetes (T1D) through immunotherapy. Strategies could include targeting specific molecules on cells that may be of importance in disease pathogenesis.
Such approaches are in progress and candidate molecular targets of potential interest have been identified. As part of the next steps, our goal is to obtain a better understanding of the relevance of candidate molecules in T1D development. For this reason, in this project we aim to study whether molecules of interest are expressed in the pancreas; the location and identity of positive cells; and the relationship to disease. This will assist in the next key stages of drug development for T1D
-

Peter Linsley & Karen Cerosaletti
Benaroya Research Institute
The role of shared germline-like TCR alpha chains of self-reactive TCRs in type 1 diabetesHuman islet antigen reactive CD4+ memory T cells (IAR T cells) play a key role in the pathogenesis of autoimmune type 1 diabetes (T1D). Using single cell RNA-sequencing (scRNA-seq) to identify T cell receptors (TCRs) in IAR T cells, we identified a multi-specific class of T1D-associated T cell receptors (TCRs) that share TCR alpha chains between individuals (“public”). We isolated IAR T cells from blood of healthy, new onset and established T1D donors using multiplexed CD154 enrichment and identified paired TCR alpha/beta sequences from 2767 individual cells. More than a quarter of TCR junctions were shared between 2 or more cells (“expanded”) and 29/47 (~62%) of expanded TCRs tested showed specificity for islet antigen epitopes. Public TCRs sharing TCR alpha junctions were most prominent in new onset T1D. Public TCR sequences were more germline-like than unique or ‘private’ TCRs, and had shorter junction sequences, suggestive of fewer random nucleotide insertions. Public TCR alpha junctions were often paired with mismatched TCR beta junctions in TCRs; remarkably, a subset of these TCRs exhibited cross-reactivity towards distinct islet antigen peptides. Our findings demonstrate a prevalent population of IAR T cells with diverse specificities determined by TCRs with restricted TCR alpha junctions and germline-constrained antigen recognition properties. Since these “innate”-like TCRs differ from previously described immunodominant TCR beta chains in autoimmunity, they have implications for fundamental studies of disease drivers, as well as translational application to the identification of immune biomarkers and targets for TCR-based therapeutics. In the present proposal, we seek to extend these findings with IAR T cells in blood to spleen.
-

Alessandra Petrelli & Anna Giovenzana
IRCCS Ospedale San Raffaele
Unraveling the Role of CD45RA+CCR7+CD4 T Cells in the Pathogenesis of Type 1 DiabetesThis study aims to investigate the role of CD45RA+CCR7+CD4 T cells in the pathogenesis of type 1 diabetes (T1D). Historically, naïve T cells were deemed irrelevant in T1D development due to their presumed lack of antigen experience. However, distinct subsets of naïve T cells with varying phenotypes and functions have been identified, with recent literature indicating that autoreactive T cells in T1D exhibit stem-like properties with phenotypes between naïve and effector/memory states.
Our data show that the enrichment of CD45RA+CCR7+CD4 T cells in the peripheral blood of newly diagnosed T1D children. Moreover, a higher frequency of this cell subset at diagnosis correlates with a poorer outcome, regardless of age. The hypothesis posits that CD45RA+CCR7+CD4 T cells form a heterogeneous pool enriched with stem-like cells possessing effector functions, actively contributing to beta cell damage and T1D development.
To investigate the heterogeneity of CD45RA+CCR7+CD4 T cells, the study will utilize single-cell RNA sequencing coupled with TCR sequencing on longitudinal blood samples from relatives of T1D patients. This approach aims to explore the phenotype and TCR repertoire, monitoring changes in plasticity and expansion of TCR clones as individuals transition through T1D stages, with special attention to stem cell memory subsets expressing CD95. Additionally, multi-color staining of pancreas sections will be performed to localize CD45RA+CCR7+CD4 T cells relative to beta cells and assess cell infiltration dynamics in different T1D stages. Pancreas tissues from non-diabetic and autoantibody-positive subjects, recent onset and long-standing T1D patients will be analyzed.
Overall, this project seeks to shed light on the potential role of CD45RA+CCR7+CD4 T cells in beta cell damage, highlighting their potential significance as overlooked players in T1D pathogenesis and contributing to the decline of beta cell function. This project is under discussion for funding from Helmsley Charitable Trust.
-

Helena Reijonen
City of Hope
Correlation of Islet Autoantigen-Specific T-Cell Repertoire in the Pancreatic Lymph Nodes and Peripheral Blood in T1D AutoimmunityIslet autoantigen specific T cells can be found in both T1D patients and healthy individuals. However, these T cells with the same antigen specificity are expected to differ in their phenotypic characteristics. We will investigate CD4 and CD8 T cells in nPOD samples from T1D patients, prediabetic and healthy subjects allowing us to compare T cell specificities and phenotypes at different stages of the disease. We have generated tetramers representing the most common HLA-DR types associated with T1D risk (DR401, 404 and 405, 301, DQ8) or protection (DR1501, DRB5 0101 and DQ 0602) containing immunodominant peptides from the major diabetes autoantigens GAD65, Proinsulin/insulin, IA2 and Zn-T8. The approach is based on the utilization of tetramers to identify and isolate autoreactive T cells from pancreatic lymph node tissue, peripheral blood (and possibly bone marrow) offers an advantage that phenotypic analyses are performed on T cells responding to a single, known antigen mediated with a defined MHC restriction. Tetramer-based technology also allows determination of the functional phenotype of autoantigen specific T cells, for example memory, avidity/affinity, cytokine profiles, homing properties, and these cells can be sorted for gene expression studies and TcR analysis. In the cases when both peripheral blood lymphocyte and pancreatic lymph node samples are available the proposed study would provide valuable information on the predictive value of blood samples as biomarker in the evaluation of the disease process. Collaboration with other nPOD investigators allows our project to generate new knowledge on the key features and the population dynamics of CD4+ T cells directly associated with islet autoimmunity and beta-cell biology in human diabetes.
-

Gary Reynolds & Alexandra-Chloe Villani
Massachusetts General Hospital
Funded by: Breakthrough T1D
Understanding common tissue tolerance mechanisms in autoimmunityA curious feature of type I diabetes (T1D) is that, while the proteins that the immune system targets are broadly expressed in beta cells, inflammation of insulin-producing cells (insulitis) is patchy and damage develops gradually. In stage 1 or 2 T1D, prior to the onset of clinically evidence disease, the majority of insulin-producing cells are spared. Following the onset of T1D, and for many years after diagnosis, most individuals still have residual uninflamed and functional beta cells. These findings are suggestive of immune mechanisms that are active in the vicinity of unaffected islets that protects them from autoreactive damage, but these are not well understood. They are of significant interest however, as a critical need in type I diabetes (T1D) is to develop therapies that can prevent progression of pre-clinical disease to overt diabetes or protect residual beta cell function. We hypothesize that, if we can identify the cellular and molecular mechanisms that constrain excessive auto-reactive responses in the pancreas, we can use this knowledge to develop therapies that enhance them.
To understand these mechanisms we propose two complementary approaches. Firstly we will use single nuclear RNA sequencing (Flex Gene Expression) to understand the cellular composition and expression profile of the pancreas at different stages of disease. Secondly we will apply spatial transcriptomics to determine how these vary in relation to their physical location and, in particular, how they vary between inflamed and unaffected islets within the same pancreas.
-
Teresa Rodriguez-Calvo
Helmholtz Zentrum München
Targeting the innate immune system to discover new therapeutic pathways for the prevention of type 1 diabetesType 1 diabetes (T1D) is an autoimmune disease, where immune cells (T cells) of the body attack and destroy the insulin-producing beta cells. T1D is, if left untreated, a devastating and life-threatening disease and despite intensive research in the field, there are still missing pieces in the pathogenetic puzzle of the disease. With this project, we plan to focus on the innate immune system and its role in the development of T1D. Innate immune cells, mainly represented here by macrophages (ΜΦ) and dendritic cells (DCs), are among the earliest responders to inflammation, infection and stress, yet their direct implication in the development of T1D remains unknown in humans. We propose to investigate the type, distribution, and main function of these innate immune cells in the human pancreas, by defining them based on their unique markers.
By the end of this project, we expect to define the innate immune cell landscape and the changes that might take place as T1D progresses. Furthermore we will use specialized in vitro experiments, live cell- and 3D-imaging as well as super resolution microscopy, and transcriptomic and proteomic assays to study islet-immune cell interactions and beta cell function. Our goal is to better define the role of innate immune cells in the pathogenesis of T1D by acquiring quantitative data from human pancreata. In addition, we aim to obtain mechanistic information from in vitro experiments to understand how innate immune cells interact with beta cells. This will allow us to identify new “players” in the pathogenesis of T1D that will serve as targets for pharmacological intervention, bringing us closer to the prevention and cure of T1D.
-

Andrea Schietinger
Memorial Sloan Kettering Cancer Center
Funded by: NIDDK
Decoding and Reprogramming Stem-Like Progenitor CD8 T cells Driving Type 1 DiabetesThe pathogenesis of T1D is complex and involves many distinct immune populations. Autoreactive CD8 T cells play a major role as the ultimate mediators of beta cell destruction in the pancreas, leading to the loss of glucose homeostasis. However, many aspects of the programming and regulation of CD8 T cells mediating autoimmunity remain enigmatic. Utilizing the clinically relevant non-obese diabetic (NOD) mouse model of T1D, we identified two phenotypically and transcriptionally distinct beta cell-specific CD8 T cell populations in the pancreatic lymph node and pancreas with distinct functional roles in driving T1D in the NOD mouse. We propose to use T1D patient samples to investigate whether similar beta cell-specific CD8 T cell populations play a role in human disease using a multidimensional approach including multi-parameter flow cytometric phenotypic analyses, bulk and single-cell RNA-SEQ (and if enough material available also ATAC-SEQ). These studies will provide crucial insights into the little-understood programming and transcriptional regulation of CD8 T cells driving beta cell destruction and could identify novel targets for the treatment or prevention of T1D.
-

Mia Smith & Peter Gottlieb
University of Colorado
Mapping islet B cells in the pancreas on a single-cell levelRecent evidence suggests a role for B cells in the pathogenesis of type 1 diabetes (T1D), particularly in individuals who develop T1D at a younger age and demonstrate rapid progression. However, little is known regarding the specificity, phenotype, and function of B cells in young-onset T1D. Recently we performed a cross-sectional analysis comparing insulin-reactive to tetanus-reactive B cells in the blood of newly-diagnosed T1D and controls using mass cytometry. Unsupervised clustering revealed the existence of a highly activated B cell subset, we term BND2, that falls within the previously defined anergic (unresponsive) BND subset. We found a specific increase in the frequency of insulin-reactive BND2 cells in the blood of young-onset T1D donors, which was further enriched in the pancreatic lymph nodes of T1D donors. We demonstrated BND2 cells are pre-plasma cells and can likely act as antigen-presenting cells to T cells. These findings suggest that activation of insulin-reactive anergic B cells likely plays a role in the rapid progression of young-onset T1D. Moreover, similar to previous findings by the Noel Morgan group, we have also identified an increase in number of B cells in the pancreas of young-onset T1D donors compared to older onset donors. However, it remains unknown what the phenotype and function of these B cells are within the pancreas. In this proposal, we will analyze the phenotype of total B cells, including whether BND2 cells are present in the pancreas, their activation status, location in relation to other immune cells, and islet health in the pancreas of young-onset versus older-onset T1D and non-diabetic organ donors using the high dimensional imaging platform, Multiplex Ion Beam Imaging (MIBI). The potential impact of these studies lies in identification of the pathogenic B cell(s) responsible for the rapid progression of disease, which will inform our understanding of the aggressiveness of young onset T1D and increase the precision of future age appropriate therapeutics.
-

Luc Teyton
The Scripps Research Institute
Funded by: NIH/NIAID, NIH/NIDDK
Analysis of Immunocytes in Human Islets of Type 1 Diabetes PatientsCD4+, CD8+ T cells and B cells will be isolated from islets of patients suffering from type 1 diabetes. Laser dissection will be used to localize and isolate single cells after tissue staining and confocal imaging at high resolution. T cell receptors and B cell receptors will be isolated from single cells and expressed to characterize antigen specificity. T cells will also be characterized for their expression profiles of cytokines, chemokines, co-receptors, and transcription factors. These studies are aimed at discovering important new antigenic targets in type 1 diabetes and understanding the pathogenic process leading to T1D.
-

Hubert Tse
University of Alabama-Birmingham
Funded by: NIH/NIDDK
Redox regulation of anti-viral responses in type 1 diabetesType 1 diabetes (T1D) is a T cell-mediated autoimmune disease resulting in the destruction of pancreatic beta-cells. Infiltrating leukocytes generate an inflammatory environment consisting of reactive oxygen species (ROS) and proinflammatory cytokines that collectively participate in beta-cell destruction and enhance the adaptive immune effector response of islet-specific T cells. Environmental factors that include viral infections are hypothesized to directly lyse pancreatic beta-cells and generate a pro-inflammatory milieu necessary to enhance T cell autoreactivity. Currently, a gap exists in our understanding of why the immune system attacks itself in Type 1 diabetics and the signals involved in initiating immune system activation in T1D. Our previous work has demonstrated the importance of oxidative stress and the generation of ROS as essential to the pathogenesis of T1D. ROS production is required for autoimmune responses during the development of T1D in Non-Obese Diabetic (NOD) mice, a mouse model that is widely used to study T1D. Mice deficient in superoxide synthesis (NOD.Ncf1m1J) are resistant to the development of T1D and our proposed studies to further define the synergy of ROS on immune responses have the potential to provide novel insight on the mechanisms associated with pancreatic â-cell destruction in T1D. Our proposed studies will test the hypothesis that ROS synthesis is necessary for efficient anti-viral responses in T1D. Specifically, we will define the role of redox-dependent signals involved in the activation of human innate immune receptors such as Toll-like receptor 3 (TLR3), melanoma differentiation-associated gene 5 (MDA5), and retinoic acid-inducible gene I (RIG-I) in response to Coxsackie virus infection with PBMCs from recent onset T1D and healthy patients. The studies in this proposal will further expand our knowledge of how innate immune-derived pro-inflammatory signals synergize to mediate efficient autoreactive T cell responses in Type 1 diabetes.
-
Hubert Tse
University of Alabama-Birmingham
Funded by: NIH/NIDDK
Analysis of innate immune response of myeloid cells in human Type 1 diabetesType 1 diabetes (T1D) is a multifactorial autoimmune disease that requires genetic susceptibility as well as environmental triggers to induce disease onset. Genetic association studies have implicated the IFIH1 gene, encoding for melanoma differentiation-associated protein 5 (MDA5), a cytosolic sensor of dsRNA, to be involved in T1D susceptibility and resistance. Stimulation of the MDA5 signaling pathway activates the transcription factors IRF3 and NF-κB p65, which induce Type I IFNs synthesis, which through autocrine signaling initiate an antiviral transcriptional program. Single nucleotide polymorphisms (SNPs) in the IFIH1 gene such as rs1990760 and rs3747517, which results in a non-synonymous mutation that changes alanine at position 946 to a threonine and histidine at position 843 to arginine, respectively, is highly associated with increased risk for T1D. Studies in human PBMCs and mice have demonstrated that the A946T SNP results in an increased sensitivity to viral ligands and subsequently a stronger downstream IFN response. Our central hypothesis is that T1D-associated SNPs result in an exacerbated islet-resident macrophage and dendritic cell immune response that result in an increase in Type I IFNs which contributes to the initiation of autoimmunity. This will be tested by using spatial transcriptomics and proteomics of paraffin- and OCT- embedded section and pancreas slices from healthy donors, patients with autoantibody positivity, and patients with T1D that are genotyped for IFIH1 SNPs that contribute to T1D susceptibility.
-

Stuart Weisberg & Donna Farber
Columbia University
Role of pancreas-associated myeloid cells in homeostasis and T1DThis project aims to uncover how macrophage dysregulation promotes autoimmune β – cell destruction. Our lab has identified distinct pancreatic macrophage subtypes that mediate localized immune control by regulating T cell activity. Aim 1 will use single cell sequencing to comprehensively profile pancreatic macrophage subsets and their communication with T cells during immune homeostasis and in Type 1 Diabetes (T1D). Our lab has developed and optimized methods for pancreatic macrophage and T cell purification to enable high resolution analysis using cryopreserved pancreas cell fractions from the islet lab. Ma tched p ancreas, spleen, and lymph node samples will be collected from 10 organ donors — 5 with early – stage T1D or islet autoantibodies and 5 matched controls — through established organ procurement networks (nPOD and our lab). We will define the tissue – specific macrophage states in homeostasis and T1D . Macrophage – T cell interactions will be modeled using computational tools to identify key signaling pathways distinguishing islet versus exocrine communication and homeostatic versus autoimmune states. Aim 2 will map the spatial distribution of pancreatic macrophages and their interactions with islets and T cells using multiplex imaging in donors with varying T1D status and genetic risk. Our lab has developed a custom multiplex panel for in situ labeling of pancreatic macrophage subtypes with distinct functions in controlling tissue repair and T cell activity. Formalin – fixed pancreas sections from 150 donors — including those with T1D, islet autoantibodies, and high/low T1D polygenic risk scores — will be imaged. We will use cutting edge computational spatial analysis tools to quantify the pancreatic macrophage subtypes and how T1D and T1D genetic risk factors impact their interactions with islets, exocrine pancreas and T – cells . Together, these complementary approaches will reveal the molecular and spatial dynamics of macrophage – T cell interactions in the pancreas and how they are altered in T1D, providing insight into mechanisms of autoimmunity and potential immunotherapeutic tar gets. -

Johnna Wesley, Matthias von Herrath
Novo Nordisk Research Center
Characterizing T-Cell Populations in DiabetesThis study seeks to understand the immune cells important for beta cell loss by studying the function and type of cells present in lymphoid tissue.
-

Defu Zeng & Alberto Pugliese
City of Hope
Funded by: DMRI-COH
Role of p21-activated secretory phenotype (PASP) in initiating type 1 diabetes pathogenesisType 1 diabetes (T1D) results from autoimmune insulitis destruction of insulin-secreting β cells. β cell senescence with upregulated expression of P21 has been observed in the adult NOD mice and new-onset patients. However, it remains unclear whether the beta cell senescence is an outcome of persistent insulitis, and the factors trigger insulitis remains unclear.
The initial infiltration in the islets of 3-week-old infant NOD mice was predominantly Mθ, but the mechanisms for the initial Mθ infiltration remain unclear. A recent report (Sturmlechner et al., Science2021) demonstrate that P21 activation not only results in cell-cycle arrest but also activate a large collection of genes that encoding chemokines (i.e., CXCL14) and cytokines (i.e., IL-6), called P21-activated secretory phenotype (PASP), which attract Mθ and other cell infiltration to exert immunosurveillance. It is well recognized that abnormal immune surveillance could lead to autoimmune diseases, although the mechanisms remain largely unknown. Our preliminary data with RNA-seq analysis and in vitro culture of CD45+ hematopoietic stem cell-derived immunocytes and CD45- parenchymal stem cell-derived fibroblast reticular cells (FRCs) and β cells from 3-week-old infant NOD, insulitis-resistant NOR, and non-autoimmune C57BL/6 (B6) mice indicate that exaggerated P21-activated PASP in fibroblast reticular cells (FRCs) and immunocytes (i.e., macrophages) intiates peri-insulitis and insulitis, leading to β cell senescence and destruction and full blown T1D.
Imaging mass cytometry (IMC), a powerful novel technology, allows us to analyze in situ tissue cells with more than 30 markers on the cell surface, intracellularly, and in the nucleus, and can identify cell subsets with different intracellular cytokine and chemokine profiles as well as intracellular signaling pathways and spatial cell distribution. We have developed IMC techniques in our lab and have applied IMC to study of GVHD target tissues (Kong et al: Blood, 2013) and pancreatic islet cellular analysis of murine and human islets (see preliminary data). In the current studies, we will analyze murine and human islets with IMC in couple with other functional studies to test whether exaggerated P21-activated PASP exists in the islets of new-onset T1D patients. If successful, the proposed studies will help establish a paradigm-shift concept of T1D pathogenesis.
-

Li Zhang
Indiana University
To identify T1D epitope using highly specific autoantibodiesDQ8/InsB:R3 was identified as an antigen by functional analysis of reactive T cells from peripheral blood, but direct evidence of antigenicity in islets is missing. The hypothesis is that the endogenous DQ8/InsB:R3 appear at the core organs of T1D in a disease stage-related timing. CODEX is a highly multiplexed tissue imaging platform enabling >50 markers to be simultaneously analyzed in single cells. We will use the CODEX platform to measure the expression of DQ8/InsB:R3 in different antigen-presenting cells, analyze the association of antigen with HLA-DQ and islet beta cells in the pancreas and pancreatic lymph nodes. Expression of DQ8/InsB:R3 only in patients and AAI+ individuals strongly support its pathogenic role in the development of T1D.
-

Todd Zion, Thomas Lancaster & Thillai Sathiyaseelan
Akston Biosciences
Targeting Islet-Antigen Specific B Cells for Deletion as a Means to Prevent Type 1 DiabetesType 1 Diabetes (T1D) is an autoimmune disease in which the body attacks and destroys its own insulin-producing β-cells. It affects over 3 million people in the United States. For most T1D patients, it is extremely difficult to achieve normal glycemic control even with frequent blood glucose measurements, diet control, careful determination of insulin dosage, and multiple daily insulin injections. Considering the numerous approaches to reduce or eliminate the physical, emotional, psychological and economic hardships of T1D, arresting or reversing the disease before β-cell destruction is the most ideal but difficult solution to implement.
A number of proposed therapeutics have focused on targeting and destroying the T-cells that are ultimately responsible for attacking and killing β-cells. However, in recent years a pathogenic role for B cells has also emerged. B cells can take up islet antigens, present them to helper T cells, and differentiate into antibody secreting plasma cells which enhance antigen uptake by antigen-presenting cells ultimately leading to the activation of cytotoxic T cells for β-cell destruction. A number of non-obese (NOD) diabetic mouse studies and at least one human clinical trial have demonstrated the ability to slow, arrest, or even reverse T1D through B-cell depletion. However, generalized B-cell depletion may lead to significant off-target safety issues thus restricting the allowable treatment duration and limiting its application to a small subpopulation of T1D patients for whom the risk is justified based on the anticipated benefits. An ideal product and treatment regimen would destroy only T1D specific Bcells while sparing the healthy B-cells. The development of just such a product, targeting insulin-specific B cells, is the focus of Akston Biosciences.
Novel Biomarkers
-

Mario Galgani
Consiglio Nazionale delle Ricerche
CD3+CD56+ T lymphocytes, as novel T1D biomarkerType 1 Diabetes (T1D) is a disease characterized by a lymphocyte-mediated destruction of insulin-producing beta-cells in genetically susceptible individuals. It has been demonstrated that lymphocytes with regulatory properties are essential for the maintenance of tolerance against self-tissues (pancreas) and alterations in the number and functions of these cells have been correlated with the break of immune tolerance.
We have recently shown that the frequency, in the blood, of a lymphocyte population co-expressing CD56 and CD3 molecules (here defined as CD3+CD56+ cells), represents a valuable predictive marker of T1D progression (Galgani M et al. 2013). Preliminary data show that the absolute number and percentage of CD3+CD56+ cells are significantly reduced in the blood of a large cohort of new-onset T1D subjects compared with sex and age-related healthy individuals. In addition, the frequency of CD3+CD56+ lymphocytes in the blood positively correlated with good glycaemic control and fasting C- peptide in recent-onset T1D children (see later). Finally, additional studies have been showing that CD3+CD56+ cells, isolated from healthy controls, suppress the activity of triggered lymphocytes involved in pancreas damage; of note, this activity was impaired in CD3+CD56+ cells isolated from the blood of T1D subjects.
We assume that number and function of CD3+CD56+ cells may serve as biological markers to monitor disease progression, in autoimmune diabetes. Functional defects of this “regulatory” cell subset could account for the exaggerate function of lymphocytes in T1D. Thus, the results of this proposal may pave the way to the better characterization of a novel cell population involved in the T1D pathogenesis, likely representing a useful biomarker potentially implicated in the control of the immune tolerance.
-

Emma Hamilton-Williams
University of Queensland
Funded by: Breakthrough T1D
Meta – proteomic biomarkers in stool for prediction of islet autoimmunity progressionType 1 diabetes (T1D) is increasing around the world due to unknown changes in our lifestyle. One factor that may drive this change is the bacteria in our intestines. These bacteria interact potently with our immune system and metabolism. We have shown that changes in intestinal and pancreatic proteins found in stool are associated with risk of T1D development. By integrating these data with microbial community profiling, we have shown that host proteins linked to gut barrier function and exocrine pancreas function are associated with changes in specific gut microbes. Now we plan to investigate whether changes in proteins found in stoolare associated with inflammation present in either the intestine or pancreas of individuals with islet autoimmunity.
-

Mark Huising
University of California, Davis
Funded by: Breakthrough T1D
Urocortin 3 as a sensitive marker for beta cell functionUrocortin 3 (Ucn3) is a neuropeptide that was discovered by our group (Lewis et al., 2001). Ucn3 is specifically expressed in mouse beta cells, but is in human islets also expressed by alpha cells (Li et al., 2003; van der Meulen et al., 2012). Ucn3 is a member of the same neuropeptide family as corticotropin-releasing factor (CRF) and both peptides activate related receptors; CRF predominantly activates the type 1 CRF receptor, while Ucn3 exclusively activates the type 2 CRF receptor. We have in recent years discovered that CRF and Ucn 3 both contribute to the regulation of insulin secretion and the maintenance of functional beta cell mass by virtue of locally expressed CRF receptors in mouse and human islets (Huising et al., 2010, 2011). Interestingly, Ucn3 is not detectable in the developing mouse pancreas until embryonic day 17.5 and is not expressed by all beta cells until postnatal day 14 (van der Meulen et al., 2012). This is considerably later than the onset of expression of transcription factors that mark mature beta cells, such as Pdx1, Nkx6.1 and MafA as well as markers of functional beta cell maturity, including Pcks1, Znt8 (Scl30a8) or Glut2 (Scl2a2; in mouse).
In line with findings in mouse pancreas, Ucn3 expression in human embryonic pancreas is not detectable until the end of the first trimester and trails the onset of expression of insulin and glucagon by several weeks (van der Meulen et al., 2013). However, in marked contrast to murine pancreas, where Ucn3 demonstrates exquisite selectivity for the beta cells, Ucn3 in both human and macaque (Macaca nemestrina) pancreas and in freshly isolated human donor islets is expressed in both beta and alpha cells (van der Meulen et al., 2012). Moreover, and perhaps to emphasize the species differences that exist regarding the local network of CRF-family neuropeptides, CRF expression is selective for human alpha cells and is not detected in mouse islets. In agreement to the above observations, Ucn3 in human embryonic stem cell-derived pancreatic endocrine cells is expressed relatively late and only after an extensive period of in vivo maturation in recipient mice and is then detectable in both mature beta and alpha cells (van der Meulen et al., 2012). These findings have established Ucn3 as a marker of beta cell maturity. We therefore hypothesize that Ucn3 is a biomarker of functional, mature beta cells, and is down regulated during diabetic conditions that are associated with beta cell dysfunction and dedifferentiation. Supported by nPOD, we will establish if Ucn3 expression is reduced in alpha or beta cells of T1D and T2D donors compared to non-diabetic controls.
-

Daniel Kaufman
University of California, Los Angeles
High-resolution mapping of the human pancreatic magnetic resonance landscapeMagnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) both detect signals from atomic nuclei and can be performed together in conventional clinical MRI scanners. MRI provides the anatomical information, after which MRS scanning can provide biochemical information on the constituents within a region of interest. MRS has been used in clinical settings to study metabolic changes within breast and prostate tumors, as well as changes in small molecules in the brain associated with seizures, stroke, Alzheimer’s, and Parkinson’s disease. This technology has not yet been extended to the analysis of pancreatic small molecules in healthy and diabetic states. As an initial step in this direction, we request nPOD human pancreatic samples from healthy and T2D and T1D diabetic individuals in order to study them ex vivo by high-resolution MRS (HR-MRS). This will provide a new read-out of pancreatic small molecule constituents in healthy versus diabetic states. The results will provide insights into the disease processes and complement data gathered by other technologies. Importantly, the results will identify potential clinical MRS biomarkers of ß-cell health. Future clinical MRS studies can then focus on the candidate biomarkers using MRI pulse sequences or data editing programs that will enhance the signals from candidate biomarkers. Thus, our proposed studies will provide basic information on pancreatic constituents in health and disease states, and will lay the foundation for developing MRS as a noninvasive tool to monitor ß-cell health in the clinic.
-

Thomas Linn & Sebastian Petry
Justus Liebig University
Redoxins in the pathogenesis of type 1 diabetesWe plan to quantify redoxin expression levels and the frequency and type of redoxin expressing pancreatic cells by imaging procedures established in our group. For this purpose we propose a pilot study with samples from #3 cases ideally with rapid (pre-type 1 or type 1 diabetes) or more chronic (type 2 diabetes) ongoing beta cell destruction process. To these samples we will apply well-established antibodies that were tested in our cell based models and non-diabetic human pancreas before. Thereby we will be able to identify the most obvious changes of expression of redoxins of interest with a focus on iron-sulphur containing clusters, such as glutaredoxins for the first time in human pathology. We will generate hypotheses from the collected results to perform power calculations and figure out if and how many samples are required for confirmation. Moreover, we will search for prominent associations of our findings to the medical history of the subjects, specifically to insulin reserve measured by C-peptide blood or urine concentrations whatever available.
At our place of research Giessen, Germany a human islet transplantation program was performed for over 20 years, but it was halted due to changes of national transplantation laws with the result of reduced availability of pancreas organ donors. From this period we have continuously collected and restored pancreas samples both embedded in paraffin and frozen from non-diabetic donors.
-

Roberto Mallone
INSERM Cochin Institute
Identification and characterization of Coxsackievirus-reactive CD8+ T cells in pancreatic lymph nodesDespite accumulating epidemiological and histopathological evidence for an association between Coxsackievirus B (CVB) and type 1 diabetes (T1D), a causal link is missing. Very little is known about the T-cell response mounted against CVB infection and the epitopes targeted. Moreover, the ongoing CVB vaccination trials require: 1) the development of T-cell biomarkers to follow vaccination efficacy; and 2) the exploration of potential cross-reactivities with islet antigens that may precipitate rather than prevent T1D. We identified the HLA Class I viral peptidome of CVB-infected β cells, pinpointed a highly focused response toward very few immunodominant epitopes recognized by CD8+ T cells, and the functional features of these T cells, e.g. their capacity to destroy infected beta cells.
We now aim to investigate whether these CD8+ T cells recognizing CVB peptides are also found in the pancreatic tissues targeted by islet autoimmunity, and whether they are enriched in these tissues as compared to peripheral ones. Such enrichment would lend support to the role of CVB infection in triggering islet autoimmunity.
-

Nadav Sharon
Technion, Israel Institute of Technology
Funded by: Breakthrough T1D
Discovery of novel autoantibodies for accurate diagnosis of T1DType 1 diabetes (T1D) is typically diagnosed when patients lose their ability to produce sufficient amounts of insulin. However, this is merely the final stage in a long process which involves the gradual loss of beta cell mass due to an autoimmune attack which specifically targets insulin producing beta cells in the pancreatic islets. Recently, the FDA approved Teplizumab – a drug which can delay the onset of insulin dependence by two years if administered before the typical point of diagnosis. Administrat ion of Teplizumab therefore requires prediction of imminent T1D , which is based on the appearance of autoantibodies against four different islet – associated proteins (antigens) in a person’s blood. However, the predictive value of these autoantibodies is limited, as they provide a very wide time window for disease onset, which ca n span a decade or more. The problem is even worse in adult onset T1D, where many patients do not develop autoantibodies against the known antigens. Here, we intend to improve early T1D diagnosis by expanding the repertoire of known autoantibodies. To discover novel antigens targeted by autoantibodies in early T1D, we will use spatial transcriptomics on cadaveric pancreata of patients with recent onset T1D, and sequence the antigen – recognition site in antibody producing molecules (B cell receptors – BCR) generated near their islets. We will then create synthetic antibodies with identical sequences, and use them to “fish” their cognate antigen. Ultimatel y, we will use blood samples from live donors to explore whether autoantibodies against the newly discovered antigens are unique to T1D patients, and examine their potential for accurate and reliable early diagnosis.
Novel Technologies
-

Bernd Bodenmiller, Mark Atkinson, Harry Nick, & Clive Wasserfall
University of Zurich
Highly Multiplexed Imaging Mass Cytometry of the Human Pancreas Affected by Type 1 DiabetesDespite extensive efforts, understanding of the mechanisms of type 1 diabetes (T1D) development in human patients is collectively lacking, particularly with regard to contributions of islet endocrine cells, the immune system, and their interplay in the disorder’s development. Until recently, these questions have been particularly challenging to address due to technical limitations in the number of analytes that can be simultaneously measured from a single tissue section (i.e., typically four to six markers per section by immunohistochemistry or immunofluorescence). To address this, we have established Highly Multiplexed Imaging (HMI) methodology based on CyTOF mass cytometry to examine human pancreas samples from organ donors with or at varying levels of risk for T1D as well as control donors. We hypothesize that imaging mass cytometry (IMC) characterization of islet and immune cells in the human pancreas will provide novel clues regarding: 1) the impact of pre-T1D and T1D conditions on islet cell phenotypes and 2) the local intra-islet—immune interface throughout disease progression. Specifically, metal isotope-tagged antibodies serve as reporters allowing for unprecedented multi-parameter measurements of single cells, now in the three-dimensional (3D) tissue microenvironment. At the RNA, protein and protein modification level, 30-100 markers expressed by islet and immune cells can be simultaneously analyzed from human pancreas sections at single-cell resolution in 3D across the lifespan to identify “normal” islet development, islet plasticity, cell reprograming, and pathology throughout T1D disease staging. In this study, we are examining: 1) islet endocrine constituents (i.e., alpha, beta, and delta) to potentially identify reprograming cells or to discover novel subsets within these known cell types; 2) cellular signaling networks in endocrine, acinar, and immune cells; and 3) their interactions at various stages throughout the natural history of T1D. Indeed, we have developed the histoCAT 3D reconstruction toolbox to generate 3D data from serial tissue sections with subcellular (i.e., pixel) resolution. This work is revolutionizing our ability to examine the human pancreas in health and disease and has facilitated new avenues of research with new research questions that were previously impossible. We anticipate that we will identify novel pathways disrupted in human T1D, the cellular and molecular networks altered during disease pathogenesis, and new disease-defining/staging characteristics, each of which may represent a potential target for the development of novel therapies aimed at preventing or halting the disease progression.
-

Joan Camunas-Soler
Gothenburg University
Integrating functional and transcriptomic measurements in pancreatic islets with spatial resolutionType 1 diabetes mellitus (T1D) is a complex autoimmune condition which is globally raising, it is influenced by genetic predisposition and environmental factors such as dietary changes and potential exposure to viruses or toxins. Single – cell methods have recently shown that endocrine islet cells are molecularly and functionally heterogeneous, and that cell – to – cell interactions within islets are critical in normal physiological function and in the development of disease like T1D. However, to date there are only a few approaches available to obtain accurate molecular and functional characterization of islet cells in their tissue context. In this project, we will combine spatial transcriptomics with functional imaging in pancreatic tissue slices and use this approach to gain novel insights into the physiological and genomic changes that take place in pancreatic islets during onset of T1D. We hypothesize that some of these changes are structured and dependent on the cellular microenvironment. This novel methodology will permit the characterization of β – and α – cell subpopulations in their tissue context during progression to T1D and link changes in cell functionality to their molecular profile. The identification of transcriptomic regulators of coordinated islet cell function and mechanisms of disruption during T1D onset will be used at a later stage to identify new diagnostic biomarkers of islet disruption and β – cell damage and potential therapeutic interventions. -

Shalev Itzkovitz
Weizmann Institute of Science, Israel
Transcriptional heterogeneity in the intact human pancreasPancreatic beta cells have been shown to be heterogeneous at multiple levels. However, spatially interrogating transcriptional heterogeneity in the intact tissue has been challenging. We developed an optimized protocol for single-molecule transcript imaging in the intact pancreas and used it to identify a sub-population of “extreme” beta cells with elevated mRNA levels of insulin and other secretory genes in mice. Extreme beta cells contain higher ribosomal and proinsulin content but lower levels of insulin protein in fasted states, suggesting they may be tuned for basal insulin secretion. They exhibit a distinctive intra-cellular polarization pattern, with elevated mRNA concentrations in an apical ER- enriched compartment, distinct from the localization of nascent and mature proteins. The proportion of extreme cells increases in type 2 diabetic db/db mice, potentially facilitating the required increase in basal insulin. Our results thus highlight a sub-population of beta cells that may carry distinct functional roles along physiological and pathological timescales. We would like to apply our new technology of smFISH in the intact human pancreas to assess whether similar heterogeneity of extreme and non-extreme beta cells exists in human.
-

Klaus Kaestner
University of Pennsylvania
Funded by: NIH/NIDDK
Imaging Mass Cytometry for the Analysis of the Human Diabetic PancreasBy integrating systematic views of the transcription, translation machinery at single-cell level, we intend to gain insights into the molecular changes in diabetes and in aging and provide the research community with useful database resources.
-
Sally Kent & Aldolfo Garcia-Ocaña
University of Massachusetts Chan Medical School & City of Hope
Funded by: City of Hope
De-differentiation of cryopreserved splenocytes to iPSC as an isogenic resource for differentiation to sc-islets. Addendum: De-differentiation of cryopreserved islet-derived T cell lines to iPSC as a resource for differentiation to sc-isletsIn collaboration with the New York Stem Cell Foundation (NYSCF, see letter of collaboration), we will attempt de-differentiation of cryopreserved splenocytes to iPSC: this has not been attempted before to our knowledge. If successful, these iPSC may be differentiated into sc-islets and serve as an isogenic sources of sc-islets for interactions with the islet-derived T cell lines the Kent lab has generated and banked. If successful, we will submit a full addendum to this project for the study of the interaction of isogenic iPSC-derived sc-islets and autologous islet-derived T cells. This technology may be used by other investigators in their examination of the isogenic interaction of sc-islets and immune cells.
Addendum:
In collaboration with investigators at the City of Hope, we will attempt de – differentiation of cryopreserved, islet – derived T cell lines from nPOD donors to iPSC: this has not been attempted before to our knowledge. In addition, we would transfer autologous, cryopreserved spleen cells to the investigators at COH. Primary splenocytes from the donor, previously supplied by nPOD to Dr. Kent, will be used in whole genome sequencing (WGS) to establishing the donor’s true germline reference genome in order to mon itor Identify any donor – specific or disease – related germline variants and confirm genetic stability and absence of reprogramming – associated mutations in the iPSCs. These data will be made available to nPOD. If successful, these iPSC will be differentiated into sc – islets and serve as an isogenic sources of islets for interactions with the islet – derived T cell lines the Kent lab has generated and banked. T h ese sc – islets may be used by other investigators in their examination of the interaction of sc – islets a nd immune cells. -

Sarah Kim & Martha Campbell-Thompson
University of Florida
Funded by: Breakthrough T1D & UF Informatics Institute
Deep Learning-based Analyses of Pancreatic Islet Beta-Cell Heterogeneity and MRI Pancreas Volume as Biomarkers to Improve Understanding of Type 1 Diabetes ProgressionThe overall objective of this project is to develop a deep learning based imaging analysis and informatics tool for better understanding of pathogenic mechanisms in T1D, using existing data which has been collected for 14 years by Network for Pancreatic Organ Donors with Diabetes (nPOD) study. The central hypothesis of this proposal is that modifications of the pancreatic islets due to loss of insulin-producing β-cells and infiltration of islets by CD3+T cells during the progression to T1D progression can be characterized and their heterogeneity can be quantified in pancreas whole-slide digital pathology images. Ultimately, our results will help in the rational design of T1D prevention and treatment strategies.
-

Chantal Mathieu & Constantia Gysemans
KU Leuven
Age-related heterogeneity of type 1 diabetes: a multi-omics, spatially resolved, exploration of the human pancreas at disease onsetType 1 diabetes (T1D) is a chronic autoimmune disease long thought to primarily affect children and adolescents. However, recent data suggest that nearly 70 percent of new T1D cases occur in adults. Striking differences in genetic, metabolic, and immune features as well as in diabetes management have been described between childhood- and adult-onset T1D, but many of these variances are poorly understood. An improved understanding of childhood- and adult-onset T1D heterogeneity is essential to provide optimal diabetes care but in addition, for designing novel disease-modifying therapies to prevent or arrest ongoing beta cell autoimmunity. I propose to implement the most advanced high-plex in situ assay platforms to study, at gene and protein level, the spatial context and interaction of cellular subtypes in pancreatic tissues obtained from childhood- and adult-onset T1D cases. Integrated data analyses will not only provide novel insights into precise cellular localizations but also identify cellular differentiation / activation states and neighborhoods. This will provide the best map yet of what is occurring in the pancreas at disease onset and how this differs between children and adults. Such knowledge is key to developing new treatments to cure T1D and to better define optimal treatment regimens.
Update:
Type 1 diabetes (T1D) is a chronic autoimmune disease long considered to predominantly affect children and adolescents. However, recent epidemiological data indicate that nearly 70% of new T1D cases now occur in adults. Childhood – and adult – onset T1D differ markedly in genetic risk, metabolic trajectory, immune responses, and clinical management, yet the biological basis of this heterogene ity remains poorly understood. A deeper mechanistic understanding of age – dependent disease onset is essential not only to optimize diabetes care, but also to guide the development of disease – modifying therapies aimed at preventing or halting ongoing β – cell autoimmunity. In recent years, our knowledge of pancreatic architecture in T1D has expanded rapidly through single – cell and spatial transcriptomic studies. These pioneering efforts have revealed immune infiltration patterns, β – cell stress states, and alterations in the exocrine compartment, fundamentally reshaping our view of the diseased pancreas. Nevertheless, most available studies remain limited by a strong focus on adult donors, incomplete age stratification, and restricted integration of transcriptomic and proteomi c information. Moreover, while immune infiltration is a central feature of T1D pathology, the spatial organization and functional heterogeneity of immune cell subtypes within the pancreatic microenvironment , particularly at disease onse t, remain incompletely resolved. The proposed work builds on and extends these advances by implementing state – of – the – art, high – plex in situ platforms to interrogate pancreatic tissues from both childhood – and adult – onset T1D at gene and protein level, with a specific focus on spatial context and cell – cell interactions. A particular strength of our laboratory is its recent interest in neutrophil biology and neutrophil heterogeneity in autoimmune and inflammatory settings, including T1D . This project will therefore place special emphasis on identifying neutrophil subtypes, activation states, and spatial niches within the pancreas. Importantly, this focus will be embedded within a comprehensive analysis of the broader immune landscape, including adaptive and innate immune cell populations, to ensure an integrated view of pancreatic immune architecture. By integrating high – dimensional spatial transcriptomic and proteomic data, we will move beyond static cell – type catalogs toward a contextual understanding of how immune and non – immune cell states, differentiation trajectories, and functional activation are organized within distinct spatial neighborhoods. This approach will enable identification of age – specific immune niches, including neutrophil – enriched microenvironments, as well as β – cell stress responses and stromal interactions that may explain differen ces in disease aggressiveness and progression between children and adults. Together, these integrated spatial analyses will provide the most detailed comparative map to date of pancreatic pathology at T1D onset across the lifespan. Such knowledge is essential for refining patient stratification, informing age – adapted therapeutic strategies, and identifying novel targets for interventions aimed at preventing or curing T1D. -
Teresa Rodriguez-Calvo & Matthias Mann
Helmholtz Zentrum München & Max-Planck-Institut of Biochemistry
Unbiased single cell resolution spatial proteomics: defining the antigenic landscape in the context of HLA-I expression during type 1 diabetes progressionDiabetes is a complex metabolic disease in which patients experience high blood glucose levels leading to a plethora of symptoms and long-term complications. In type 1 diabetes (T1D), this happens likely due to an autoimmune reaction targeting the insulin producing cell population, so-called beta cells, within the pancreatic islets. CD8+ T-cells are considered the main cell type implicated in beta cell destruction. However, their phenotype, recognized antigens, as well as how they interact with beta cells is still rather unknown. During disease progression, islets with a heightened expression of HLA class I (HLA-I) proteins can be observed. This feature is usually detected in all cells within one islet but not in all islets within one donor. The increased expression of HLA-I molecules on the surface of islet cells, and especially of beta cells, could be a precipitating factor and actively contribute to the presentation of self-antigens to the immune system. Based on this, we propose to investigate the differences at the proteomic level between islets with low, medium and high expression of HLA-I molecules on a cell-type resolved basis (in different cell types) using our recently published method Deep Visual Proteomics (DVP). This platform combines artificial-intelligence-driven image analysis of cellular phenotypes with automated single-cell or single-nucleus laser microdissection and ultrahigh-sensitivity mass spectrometry. Our goal is to analyze the proteomes of single-cell populations within their spatial context to provide answers regarding the significance of HLA-I upregulation with respect to disease etiology, as well as the proteomic profile of different islet cells during disease progression.
-

Maike Sander & Chun Zeng
Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Germany
Funded by: NIH-NIDDK (HIRN)
Single cell analysis of the human pancreas in type 1 diabetesType 1 diabetes (T1D) is an autoimmune disorder that affects 1.25 million individuals in the US and for which there is no known cause or cure. T1D is characterized by autoimmune destruction of insulin- producing beta cells in the pancreatic islets. The pancreas consists of multiple different cell-types relevant to diabetes pathogenesis including endocrine (beta, alpha, delta, gamma, and epsilon), exocrine (ductal and acinar), immune (macrophage, dendritic, T cells, and others), and endothelial cells. In addition, there is evidence for further cellular heterogeneity within individual cell types. Each cell- type has distinct functions in pancreas biology, yet the complete repertoire of pancreatic cell-types in non-diabetic and diabetic individuals, and the regulatory programs that define their identity and their roles in diabetes pathogenesis are unknown. Technological advances have enabled profiling chromatin and gene expression in individual cells which can reveal regulatory programs in specific cell types to facilitate understanding their role in disease. For this project we will adapt cutting edge single-cell methods on nPOD frozen samples from non-diabetic and diabetic donors to define cell-types and identify mechanisms of disease promoting cross-talk between cells.
Pathology
-

Joana Almaça
University of Miami
Funded by: NIH-NIDDK (HIRN)
Changes in human islet microvasculature during the progression of type 1 diabetesEndocrine cells in pancreatic islets are embedded in a network of microvessels that regulate islet blood flow and hormone secretion. It is still unknown what happens to the islet microvasculature in type 1 diabetes (T1D), despite being the site of immune cell recruitment and infiltration. The microvasculature consists of capillary tubes made of a thin layer of endothelial cells covered by pericytes. Pericytes are the mural cells of the microcirculation. They are crucial for proper microvascular function, by giving structural stability, participating in angiogenesis, controlling vascular permeability and blood flow. While a wide body of literature exists on islet endothelial cells, the function of the islet pericyte is largely unknown. The aim of this research project is to look at pericytes in islets from non-diabetic donors as well as in individuals at different stages of type 1 diabetes. My hypothesis is that loss of pericyte coverage of capillaries leads to microvascular dysfunction and immune cell infiltration, contributing to the development of T1D. I also plan to assess whether pericytes control human islet capillary diameter, similarly to their role in mouse islets.
-

Joana Almaça
University of Miami
Changes in pericyte phenotype and function during type 1 diabetesEndocrine cells in pancreatic islets are embedded in a network of blood vessels that support adequate nutrient sensing, efficient release of hormones, and timely responses to changes in glycemia. This network of islet blood vessels consists of capillary tubes made of endothelial cells that are covered by pericytes. Compelling evidence indicates that significant structural alterations of islet blood vessels occur in individuals with T1D. These include not only changes in blood vessel diameter and density, altered expression of adhesion molecules by endothelial cells, but also perivascular alterations such as increased infiltration of immune cells, and altered composition of the extracellular matrix. Pericytes are part of the pancreatic islet vascular niche but it is not known what happens to them during T1D progression. The aim of this research project is to investigate pericytes and examine their phenotype and response profile in islets from non-diabetic donors as well as in individuals at different stages of T1D. Based on recent preliminary data obtained using nPOD material (under the parent project; see below), our hypothesis is that islet pericyte dysfunction early on in the disease impairs microvascular function, favoring immune cell infiltration and contributing to the development of T1D.
-
Joana Almaça
University of Miami Miller School of Medicine
Role of pericytes in innate immune responses in pancreatic isletsVascular defects are present in islets during stages 1 and 2 of type 1 diabetes, when symptomatic disease has not yet developed. However, the cellular and molecular mechanisms underlying vascular dysfunction and they link with autoimmunity have not been identified. This knowledge is crucial to understand T1D pathogenesis. In this proposal we plan to start exploring the potential involvement of pericytes in innate immune respons es in islets and their functional crosstalk with macrophages in physiological and pathophysiological states. These are highly innovative topics, crucial for our understanding of how vascular homeostasis is maintained in islets and how it is disrupted in di sease. Mechanistically, we plan to examine purinergic signaling in vascular and peri – vascular cells in islets at different stages of the disease. Because islet vascular alterations start in pre – symptomatic stages, elucidating the mechanisms of vascular dys function can shed new light into our understanding of T1D pathogenesis, and potentially reveal novel pathways/cellular players which could be targeted to delay/prevent onset. -

Alejandro Caicedo
University of Miami
Funded by: NIH/NIDDK
The sensory innervation of the pancreatic islet in health and diabetesThe role the nervous system plays in modulating islet inflammation and diabetes pathogenesis has barely been examined. Recent papers suggest that changes in sensory innervation initiate autoimmune diabetes in mice, which is in line with the notion that sensory neurons contribute to inflammation in several chronic disorders. Thus, the local interactions between the immune system and nerves in the islet may be crucial for the progression of diabetes. We propose that pathological activation of sensory nerves creates a chronic inflammatory milieu in the islet that ultimately leads to autoimmunity in susceptible individuals. Excessive activation of sensory nerves, as it may occur during hyperglycemic stress or infection with diabetogenic viruses, may lead to deranged secretion of neurotransmitters that alters vascular and macrophage functions, promoting inflammation. Studies, however, are limited by the difficulty to visualize and manipulate these processes in vivo and longitudinally in the hardly accessible pancreatic islets. Here we propose to identify the pattern of sensory innervation of the pancreatic islet in health and diabetes, using techniques our laboratory developed recently. At the successful completion, this project will provide fundamental knowledge about how the sensory innervation of the islet changes during diabetes progression. This in turn will provide the basis for functional and interventional studies to understand the role of sensory nerves in diabetes pathogenesis.
-

Nikki Farnsworth
Colorado School of Mines
Funded by: American Diabetes Association
The role of the pancreas microenvironment in type 1 diabetes progressionThe pancreatic islet is surrounded by an extracellular matrix (ECM) composed of laminin, collagen IV, and proteoglycans, that support islet function and viability. Type 1 diabetes (T1D) is characterized by the immunemediated loss of insulin-producing β-cells in the islet. Islet infiltration by activated immune cells results in the degradation of the peri-islet ECM eliminating critical signaling pathways for islet function and survival. The peri-islet ECM may provide a critical barrier to islet infiltration; however, the cellular and molecular contributions to loss of the peri-islet ECM are not well studied, limiting potential therapies targeted at this critical step in T1D. Furthermore, understanding the mechanisms underlying ECM regulation of islet function and how loss of these interactions affects β-cell survival in T1D is critical to understanding and halting T1D pathogenesis. The goal of this study is to determine the respective contributions of immune cells and β-cells to loss of the peri-islet ECM and determine how changes in the peri-islet ECM impact β-cell survival in the context of T1D. We propose 2 aims to achieve this goal: 1) Determine the specific contributions of CD4+ and CD8+ immune cells in degradation of the peri-islet ECM and loss of β-cell mass ECM in T1D, and 2) Determine the contributions of stressed β-cells in degradation of the peri-islet ECM and loss of β-cell mass ECM in T1D. Experiments for both aims will utilize human pancreas sections from the healthy controls, auto-antibody positive donors at risk of developing T1D, and donors with established T1D. The results from this project will define a new for changes in the peri-islet ECM in contributing to decreased islet survival in T1D and will provide novel therapeutic targets to halt immune infiltration of the islet and improve islet survival in T1D.
-

Rebecca Hull-Meichle & Sakeneh Zraika
University of Alberta, Canada
Funded by: Cystic Fibrosis Foundation, NIDDK Cystic Fibrosis Research Translation Center
Islet Pathology in Cystic FibrosisInsulin deficiency is a common feature of cystic fibrosis (CF), resulting in CF-related diabetes (CFRD) in ~20% of adolescents and ~50% of adults with the disease. The clinical consequences of insulin deficiency in CF are significant – CFRD is associated with worsened CF lung disease and increased morbidity and mortality. The loss of exocrine pancreatic tissue in CF is extensive, with its destruction being initiated around/before birth and resulting in pancreatic insufficiency in 85% of CF patients. In contrast, while most studies describe some loss of β cells, islets generally survive. This suggests that enhancing endogenous insulin secretion may be a viable therapeutic strategy in CF. However, the mechanisms underlying impaired insulin release in CF are poorly understood.
-

Marcia McInerney
University of Toledo
Insulin Receptor on T cells in T1DT cell movement into the islets in the pancreas, is a hallmark feature of Type 1 diabetes (T1D) that has been demonstrated on autopsy in human pancreas specimens and in mouse models of T1D such as the non-obese diabetic (NOD) mouse3-8. By determiningthe role that insulin receptor (IR)plays in this movement of the T cells may generate anew immunotherapeutic intervention for T1D, especially since chemotactic signaling and metabolic signaling are mediated by distinct parts of the IR9. Blocking IR mediated chemotaxis without affecting insulin binding and signaling would offer a new pharmacotherapeutic target, since it would effectively block T cell movement into the pancreas. Blocking T cell movement would stop or delay diabetes development in diabetes prone individuals and may allow for beta cell regeneration. Chemotaxis to the islets via IR expression is the hypothesis. To investigate this, we will examine T cells in human islets (control and T1D) for IR expression.
-

Jonathan Sweedler
University of Illinois at Urbana - Champaign
Funded by: American Diabetes Association
Understanding diabetes progression via tissue chemical imagingType 1 diabetes (T1D) is a devastating multifactorial disorder manifested by the autoimmune destruction of insulin-producing beta cells leading to insulin deficiency. Our overarching aim is to investigate the spaciochemical changes in the human exocrine and endocrine pancreata during various stages of T1D with unmatched chemical specificity and multiplexity. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry imaging (MSI) will be used for multiplexed direct measurement of metabolites, lipids and peptides in control, no diabetes autoantibody positive and affected by clinically diagnosed T1D pancreata sections. Two groups of T1D affected donors will be investigated as well as no diabetes autoantibody positive group and compared to control group and group developed T2D. Donors will be similar to ones described in 1. The first will be those with less than one year since disorder diagnosis and the second with more than 7 years since diagnosis. C-peptide levels have to drop approximately 10 times for each group with most advanced T1D cases exhibiting lowest concentrations. We will perform a comparative analysis of the pancreas head and pancreas body regions which demonstrate different extent of damage during T1D progression.1 Also, for comparative analysis of malfunctioning pancreas biochemistry, we are requesting sections of late stage T2D-affected pancreata. This work will provide direct information on the multiplexed chemical heterogeneity and plasticity in the exocrine and endocrine tissues of the pancreatic head and body in health and disorder. This effort involves new and innovative measurement technologies including ones developed in collaboration with the nPOD2 to obtain information on metabolite, lipid, and peptides. Including their spatial distributions; these data cannot be easily obtained by other methods. This research will improve our understanding of the changes of tissue surrounding beta cell destruction in T1D by elucidating the chemical signals characteristic of T1D in comparison with control and T2D, and may help to identify novel factors and pathways for therapeutic intervention to treat this disease.
-

Bruce Verchere
University of British Columbia
Funded by: Breakthrough T1D/CIHR
IAPP and innate immunity in T1DIslet amyloid polypeptide (IAPP) is a beta-cell peptide hormone that forms toxic and pro-inflammatory aggregates in pathological states, including T2D and islet transplants. Recent evidence suggests the presence of fibrillar aggregates of IAPP (islet amyloid) in some T1D pancreatic islets. In T2D and mouse
models of islet amyloid formation, aggregates of human IAPP recruit and induce a pro-inflammatory phenotype in macrophages in pancreatic islets. Since islet macrophages may play a key role in triggering autoimmunity in T1D, it follows that IAPP aggregation in stressed islets could be a key initiating event in T1D, via induction of inflammation and islet macrophage activation. Here we will study nPOD pancreas sections for the presence of pre-fibrillar (oligomeric) IAPP aggregates and fibrillar amyloid, the association of IAPP aggregates/amyloid with islet macrophages, and the presence of markers of pro-inflammatory macrophage phenotype. The goal is to determine if induction of islet inflammation induced by IAPP aggregates may be an early event in T1D.
Type 1 Diabetes Etiology & Environment
-

Mark Atkinson, Irina Kusmartseva, Clive Wasserfall, Michael Haller, Martha Campbell-Thomspon, Bernd Bodenmiller, Stephan Speier & Alberto Pugliese
University of Florida
Analysis of major digestive enzymes expression in human pancreasType I diabetes is characterized by significant loss of beta cells and decrease in pancreatic weight. It is widely accepted that decline of pancreatic weight is attributed to acinar atrophy which in turn could lead to pancreatic exocrine insufficiency frequently found in diabetic patients. However, the fact that the exocrine pancreas is affected in patients with T1D diabetes has often been overlooked. Thus, as a basis for a better understanding of pancreatic exocrine function, we propose to investigate the expression pattern of major digestive enzymes in the pancreas of nondiabetic, autoantibody positive without diabetes and T1 diabetes nPOD donors. This will be achieved by multiplex immunohistochemical staining for amylase, lipase and trypsinogen (amongst others) and by employing fresh tissue slicing technique to obtain functional readouts of pancreatic endocrine and exocrine cell function.
-
Anil Bhushan and Seung Kim
University of California, San Francisco
Stromal sentinel cells are activated in T1D and organize tertiary lymphoid structuresPreliminary data indicates that stromal cells within the vicinity of islets acquire properties that can organize ectopic lymph node like structures (known as tertiary lymphoid structures or TLS) in NOD mice. These properties include differentiating of stromal cells into specific cell types that can secrete chemokines to organize infiltration of T and B cells and secrete growth factors to organize endothelial cells into venules through which these lymphocytes traffic. These ectopic lymph nodes correlate with disease progression and mice that don’t develop disease do not show an evidence of these TLS. In this application, we utilize Co-Detection-by-InDEXing (CODEX), a multiplexed single-cell imaging technology that incorporates DNA barcoded antibodies to visualize 100 + cellular markers at the single-cell level to map islet stromal cells and TLS in nPOD samples to map tissue architecture and characterize stromal-immune interactions in T1D. As TLS have been recently been shown to be a feature of T1D pancreas in humans, we would like to assess whether the stromal cells in nPOD donors acquire TLS organizing properties. We also assess the subtypes of stromal cells with lymphoid organizing properties and expression of cell type specific markers of stromal cell subtypes involved in TLS organization. We have built our CODEX panel to include markers of lymphoid organizing stromal cells that will enable us to identify the sub-types of stromal cells. The CODEX panel includes markers for T cells, B cells and endothelial structures called high endothelial venules (HEVs). We will also be able to assess the activation state of lymphocytes. Finally we have markers for endocrine cells. We envision that such analysis of T1D samples will allow us to establish whether stromal cells are strategically positioned in the islet to provide a scaffold and orchestrate TLS formation by modulating immune cell maturation, migration and activation.
-
Alejandro Caicedo
University of Miami Miller School of Medicine
Harnessing the local cholinergic anti-inflammatory reflex in the pancreatic islet to delay or prevent type 1 diabetesType 1 Diabetes (T1D) is characterized by autoimmune destruction of insulin-producing beta cells within the pancreatic islets. Resident macrophages in the islets play a key role in maintaining tissue homeostasis but can also promote inflammation. The cholinergic anti-inflammatory reflex, known to suppress inflammation, may influence islet macrophages, but direct cholinergic innervation has not been established. This project aims to determine whether local cholinergic innervation suppresses islet macrophage activation.
Based on preliminary data showing that (a) islet macrophages are densely innervated by cholinergic nerves and (b) cholinergic stimuli dampen macrophage responses to ATP in living pancreatic slices, we hypothesize that cholinergic input prevents islet macrophages from transcribing proinflammatory cytokines through stimulation of nicotinic acetylcholine receptors. Using technological platforms we developed to investigate macrophages and stimulate local cholinergic neurons in the pancreas in situ we will pursue two aims: (1) Determine the immunomodulatory effects of cholinergic input on islet macrophages in the mouse and human pancreas and (2) Determine anatomical and functional changes in cholinergic control of islet macrophages in living human pancreatic slices at different stages of islet autoimmunity. Although macrophages are thought to play an important role in autoimmune initiation of T1D, we do not know how neural control of this cell population changes during T1D development. For these reasons, we will compare macrophage responses to cholinergic modulators in islets from Aab−, non-diabetic (control) donors with those in single Aab+ (GADA+; “stage 0”) and recent onset T1D organ donors (stage 3) using time-lapse confocal microscopy and living pancreas slices.
We anticipate completion of this project to provide necessary knowledge for future clinical interventions based on vagus nerve electrical stimulation for treating diabetes. In future proposals we will extend these studies to investigate cholinergic modulation of islet macrophages in living human tissue slices.
-

Clive Wasserfall, Mark Atkinson & Harry Nick
University of Florida
Role of Monogenic Diabetes, Integrated Stress Response, mTOR and NR4A genes in Type 1 DiabetesTo date, over 60 loci have been identified, including the HLA and insulin regions and various single nucleotide polymorphisms (SNPs), conferring genetic susceptibility for type 1 diabetes (T1D); however, a number of these loci do not correspond to specific genes and as such, do not infer disease mechanisms or etiology. We have taken an innovative strategy to identify genes/biomarkers functionally and phenotypically associated with autoantibody positivity (AAB+, considered pre-T1D) and T1D, exploring early T1D pathogenic determinants in human pancreas. Our preliminary data demonstrate that genes associated with monogenic diabetes, which result from Mendelian inherited single gene mutations and are intrinsically non-redundant, display altered expression in pancreata from AAB+ and/or polygenic T1D. Using RTqPCR on total RNA from human pancreata from the Network for Pancreatic Organ donors with Diabetes (nPOD), we studied expression of 42 monogenic diabetes genes. We identified 19 genes induced in T1D versus control donor pancreata. 8 of the induced genes were functionally linked to the unfolded protein response (UPR): EIF2AK3 (PERK), head of one of three canonical UPR branches, exhibited the highest induction in T1D pancreata. Our data strongly argue that the T1D pancreas exhibits an integrated stress response (ISR), potentially in response to pathological challenges. Given that other stresses can activate the ISR, also regulating protein synthesis and cell viability, we will investigate pathways with logical overlap, namely those associated with mTOR (mammalian target of rapamycin) and NR4A (nuclear orphan receptor) and these test the hypothesis that differential gene expression in human pancreas and blood represent prognostic, diagnostic and therapeutic biomarkers/targets in T1D or pre-T1D.
-

Knut Dahl-Jorgensen & Lars Krogvold
Oslo University Hospital & University of Oslo
Funded by: European Union's Seventh Framework Programme
Diabetes Virus Detection Study (DiViD)The Diabetes Virus Detection Study (DiViD) is the first to examine fresh pancreatic tissue at the diagnosis of type 1 diabetes (T1D) for the presence of viruses. Surgical minimal pancreatic tail resection was performed in general anesthesia 3-9 weeks after onset of type 1 diabetes in six adult patients (age 24-35 years). Pancreatic tissues were snap-frozen within three minutes after dissection, live tissues collected for functional studies, and tissues fixated with formalin and glutaraldehyde and stored. Duodenal biopsies were collected by gastroscopy. Serum, PBMCs and serum/plasma were also collected. All patients had HLA-risk for T1D and at least one positive autoantibody. All six cases fulfilled the criteria for insulitis (5-58% of the insulin-containing islets in the six patients were affected by ≥15 T cells). We demonstrated a low-grade, persistent enterovirus infection in the pancreatic islets of Langerhans. Both viral genome and viral capsid protein was detected and confirmed in different laboratories. We also observed a reserve reservoir of betacells in the islets that improved insulin secretion in a non-diabetogenic environment in vitro. This reservoir may restore the need for insulin if the disease process could be halted at the time of diagnosis. Recent gene expression studies of laser-captured pancreatic islets from the same DiViD patients show significant enrichment of gene ontologies for viral and reproduction/infectious cycle, and also significant mitochondrial dysfunction and increased cellular endoplasmatic reticulum (ER) stress. Pancreas samples from the nPOD program, recovered from organ donors, will be used as a control group together with the data generated from the biopsy of the patients from the DiViD study.
-

Tina Fløyel
Steno Diabetes Center Copenhagen
Dysregulation of molecular and cellular pathways in the pancreas during type 1 diabetesType 1 diabetes (T1D) is an autoimmune disease in which the insulin – producing β cells in the pancreas are selectively destroyed. Both genetic and environmental factors , such as viral infections, are implicated in the disease pathogenesis but how these factors contribute to the selective destruction of β cell s at the cellular level remains unclear . Accumulating data indicate that innate antiviral immune pathways (InAIPs), lysosomal cathepsin proteases, and T1D susceptibility genes play central roles in the development and progression of T1D . Genetic association studies have identified more than 140 genetic variants that influence T1D risk , incl uding several risk variants localized to InAIP and cathepsin genes. Previous and preliminary studies of laser – dissected pancreatic islets ha ve revealed differentially expressed InAIP and cathepsin genes in individuals with T1D compared to healthy individuals. However, the specific islet cells driving these changes and how they relate to disease stage or viral presence remain unknown , mainly due to the lack of spatially resolved data in human pancreatic tissue. Using advanced spatial technologies, t his project aims to pinpoint and investigate dys regulated genes, and their associated molecular and cellular pathways , involved in T1D pathogenesis , with emphasis on how InAIPs , cathepsin s , and T1D susceptibility genes might act as biomarkers and/or mechanistic drivers of β – cell dysfunction and damage in T1D. S patial transcriptomics (Xenium) and spatial proteomics (MACSima) will be performed on neighbouring pancreatic tissue sections from healthy donors, autoantibody – positive (a b+ ) donors , and donors with new – onset T1D (≤ 6 months after disease onset ) . These analyses will reveal how disease – relevant genes and proteins are expressed in the different cell types within the pancreatic islets and surrounding tissue during the early disease stages , and how their spatial distribution relates to immune cell infiltration , viral infection and β – cell destruction . This will provide knowledge about their potential mechanistic roles in T1D, with the ultimate goal of identifying novel biomarkers of early T1D and therapeutic targets for future disease prevention and intervention strategies. -
Herbert Gaisano
University of Toronto
Funded by: Breakthrough T1D/NIH
Assessing T1D islet dysfunction: β-β cell connectivity defects, T-cell induced islet dysfunction, and cystic fibrosis-related diabetes (CFRD)The availability of pancreas slices from nPOD from T1D and normal control donors has provided a huge thrust in the study of T1D islet biology as it requires the inflamed islet to be intact within a pancreas slice for accurate assessment. We have developed capabilities to culture these pancreas slices for ~10 days allowing virus gene transfer of organelle-specific fluorophores and perform live-cell imaging to track islet cell secretory function (calcium, exocytosis) and potentially other organelle disruption (mitochondria, autophagy). We plan to use these capabilities on the following 3 aims.
Aim 1: Assess β-β cell and α-α cell connectivity that we hypothesize to be differentially perturbed between early onset Aab+ islets vs late-stage T1D, which can disrupt their secretory function; and test potential agents that can rectify these connectivity defects and improve regulation of secretion. β-cells and α-cells will have cell-targeted expression of a calcium and exocytosis fluorophores as reporters for secretory function.
Aim 2: Assess how engineered human T cells (Tregs, CD4 and CD8) that interact with islets (β-cell and α-cell) to cause initial disruption of function progressing to β-cell killing and α−cell dysfunction (causing glucose blindness). Tcells will be tagged with red fluorophore and β- (or α-) cell will be tagged with green fluorophores to visualize their interactions in real time and at high spatial resolution. This is a collaboration with KQ5 group – Qizhi Tang.
Aim 3. Assess how pancreatic duct defects or deficiency of CFTR cause islet dysfunction. We will knockdown CFTR expression with an adenovirus in human pancreas slices and assess the islet cell function by functional imaging as described above. This is in collaboration with KQ3: role of exocrine pancreas in T1D; and is in collaboration with Rebecca Hull (Seattle and Edmonton).
-

Dirk Homann & Mihaela Stefan
Icahn School of Medicine at Mount Sinai
Role of environmental chemicals in the development of type 1 diabetesThere is a well-documented rise in the incidence of type 1 diabetes (T1D) in industrialized countries suggesting a role for environmental chemical exposures. Alarmingly, the increased incidence is seen mostly in children under age 5. However, research on environmental chemical exposures and T1D is very limited and there is an urgent need for research to fill this gap in our knowledge. Therefore, the goal of this proposal is to identify environmental pollutants that play a role in the increased incidence of T1D. We hypothesize that in the first years of life in T1D genetically susceptible children certain pollutants trigger damages specific to beta-cells making them more susceptible to the subsequent autoimmune attack. Of particular importance is the possibility that chemicals can directly affect beta cell physiology and thus may trigger initial insults precipitating the activation of self-reactive T-cells. Therefore, we have adapted a novel multi-chemical tissue mapping technology to study chemical quantitative distributions within the pancreatic tissues from T1D patients and healthy controls. Quantitative assessment of distribution of toxic and essential metals can reveal adverse (and protective) effects that occur at the cellular levels and may be overlooked in studies measuring average concentrations in homogenized pancreatic tissue. Laser ablation-based quantitative imaging provides a powerful tool to study the role of environmental toxicants and essential elements in T1D.
-

Heikki Hyoty
University of Tampere, Finland
Funded by: Breakthrough T1D
Virus detection in pancreas and other tissuesEnterovirus infections cause pancreatitis and damage endocrine cells in the pancreatic islets. Epidemiological studies have shown an association between enterovirus infections and type 1 diabetes, and viral structures have been detected in the pancreas of diabetic patients. Our recent epidemiological studies have suggested that among all different enterovirus types, the group of coxsackie B viruses is linked to the initiation of beta-cell autoimmunity and development of type 1 diabetes. However, it is not known if this association is causal, and further studies are needed to address this important question. Current evidence suggest that enteroviruses can establish a slowly replicating persisting infection in the pancreas. In such case, the virus should be detectable by highly sensitive assays in the pancreas of diabetic and prediabetic patients. Our aim is to develop such assays, validate them in collaboration with other nPOD-V investigators and screen nPOD tissues for enteroviruses using the most optimal test panel.
Our primary aims include the development of different histological methods, as well as PCR-based techniques, for enterovirus detection in various samples. These techniques are utilized in detecting enteroviruses in various tissue samples – such as pancreas, duodenum and spleen – of T1D, pre-diabetic and non-diabetic donors, and further in comparing the prevalence of EV infections between different donor groups, as well as in different tissues. We are applying three main methods to accomplish our goals:
- Immunohistochemistry (IHC) with a wide panel of both commercial and in-house antibodies suitable for FFPE and frozen samples.
- Standardized in situ hybridization (ISH) assay suitable for FFPE and frozen samples. We are using novel probes for both general and serotype-specific detection of enteroviruses.
- Highly sensitive RT-PCR method for the detection of viral RNA and for the sequencing of viral genome.
Our studies have shown that these markers of enterovirus infection are more prevalent in type 1 diabetic and pre-diabetic subjects than in non-diabetic controls, and that the presence of enterovirus in pancreas correlates with that in other tissues.
-
Pia Leete & Joanna Boldison
University of Exeter Medical School
Funded by: RD Lawrence Fellowship Diabetes UK & Leona M. and Hary B. Helmsley Charitable Trust
The Heterogeneity of Type 1 Diabetes: An Islets StoryWhen a person is diagnosed with Type 1 diabetes (T1D), t he amount of beta – cell mass each individual lose s is highly variable despite their clinical need for insulin therapy. Attempts to detect robust markers in the blood that precisely describe disease activity in the islets are ongoing , but without a full understanding of the disease course in the pancreas, when attempting to target therapy to the organ , we are still operating par tially blindfolded. We have previously reported that two forms, or endotypes, of T1D are evident in the pancreas [1 – 4] . T hese differ according to the degree of beta – cell loss, the composition of the islet – specific immune infiltrate , and how beta cells process insulin . Additionally, we now have preliminary data that suggests the beta – c ells them se lves may vary between the two forms ) , even hint ing that successful beta – cell regeneration may be more likely in one form than the other . Our proposed study will expand on these investigations , and we will compare pancreatic protein expression profiles between people diagnosed with T1D at different ages, with different durations of disease and compare this to those without diabetes and with Type 2 diabetes (as a proxy for metabolic dysregulation) . We will leverage advanced histological and imaging techniques to analyse the presence of multiple critical proteins in granular detail in whole sections of pancreas . W e will then (b ased on several features of pancreatic T1D and beta – cell loss ) , construct a nuanced ‘pseudo’ timeline of disease progression in T1D to illustrate the life and loss of beta – cells in T1D and explo re if this is the same in all p eople, randomly different between people – or st ra tifiably different between gro ups of people living with T1D . With this information we will bett er understa nd if we should be looking for distin ct forms of T1D , and this may h elp scientists and trialist s underst and why some people re spond brilliantly in clini cal trials, whilst other do not. -
Kathrin Maedler & Federico Paroni
University of Bremen
Funded by: ERC
Detection of coxsackievirus infection in β-cells in T1D by short fluorescently labeled oligonucleotide probesType 1 diabetes (T1D) results from a complex interplay between genetic polymorphisms, immune system and environment. Viruses, mainly enteroviruses such as members of the Coxsackie B virus (CVB) family, have been suspected since long time to trigger diabetes and epidemiological studies support a causative role of viral infections during diabetes onset. Nonetheless, the presence of the enterovirus in diabetic tissue has not been fully confirmed. Here we present an adapted method to target single RNA molecules with short fluorescently labeled oligonucleotide probes in situ, that anneal to common regions of the coxsackievirus family. We successfully detect viral RNA in both cell culture and formalin fixed tissue sections bypassing the potential limitation of fragmented RNA. With the use of small contiguous RNA probes we were able to detect lower levels of strain specific infection compared to classical immunostaining. Specificity of detection was further confirmed without the well-known cross-reactivity with cellular proteins shown by antibodies. Furthermore the use of RNA probes allowed the discrimination between single RNA molecules and virus replication loci. Our data show a novel method to analyze human samples for the presence of enteroviral RNA and will be ideally suited to detect enterovirus infection with a very high sensitivity in paraffin embedded or frozen pancreatic sections.
-
Clayton Mathews & Martha Campbell-Thompson
University of Florida
Funded by: NIH-NIAID
Investigating the role of iron metabolism in beta cell function and the immunopathology of type 1 diabetesInsulin release from beta cells depends on ATP energy g enerated by mitochondrial . The electron transport chain is the primary group of proteins that make ATP energy and they require iron to do so. Consequently, iron is essential for insulin release, and iron deficiency inhibits beta cell function. Conversely, excess cellular iron leads to an incre ase in reactive oxygen species (ROS), which can damage cells and cause dysfunction . Evidence indicates that both R OS and mitochondrial dysfunction occur during diabetes progression . B eta cells have low antioxidant capacity making them susceptible to ROS from iron overload. Iron overload can cause mitochondrial fragmentation and dysfunction . Indeed, diseases of iron overload such as hereditary hemochromatosis and Friedreich’s ataxia have an associated risk of diabetes in the absence of autoimmu nity . I nflammation alters iron metabolism by promoting sequestration within cells. Prolonged inflammation can cause anemia of inflammation , which has been reported in patients with T1D, where serum iron levels are too low to promote the differentiation of red blood cells . An inflammatory signaling molecule called interferon gamma (IFNy) is increased in in dividuals who develop diabetes long before the onset of disease . IFNy has been shown to increase cellular iro n . Taken together, these data demonstrate the importan ce of understand ing iron metabolism under basal conditions and dur ing T1D disease progression in beta cells. Importantly, iron metabolism pathways may serve as targets for intervention for the treatment of diabetes. -

Noel Morgan & Sarah Richardson
University of Exeter Medical School, UK
Funded by: EU-FP7
Enteroviral infection as a causative factor in type 1 diabetesConsiderable evidence has accumulated implying that enteroviral infection is associated with the development of type 1 diabetes but the precise nature of this relationship remains unclear. Many studies have revealed that enteroviral RNA is detectable in patient serum at, or before, the onset of disease but it is unclear whether viral infection also occurs commonly in other organs and tissues at this time. We are investigating this possibility using pancreas (and other tissue) samples recovered from patients with recent-onset type 1 diabetes which are made available from within the nPOD collection. We study these samples in parallel with a larger collection available to us within the UK.
Our initial aims are to evaluate whether enteroviral infection can be detected at the level of the pancreas in patients with type 1 diabetes and to compare the prevalence with relevant age-matched controls. To achieve this, we are employing immunological approaches using antisera raised against enteroviral capsid proteins to detect infected cells and to provide unambiguous identification of these cells. Such studies are coupled to parallel experiments designed to define whether a “viral footprint” can also be found which would indicate that infected cells mount an active anti-viral response. Among the candidate molecules comprising such a “footprint” we are examining relevant pathogen recognition receptors, pro-and anti-apoptotic proteins, interferon response genes and downstream targets of interferon signalling.
A further goal is to establish whether the infection of islet cells by enteroviruses follows an atypical course in type 1 diabetes. In particular, we are developing methods to differentiate between more classical acute infections (which culminate in cell lysis) and those which persist for prolonged periods without causing the demise of the cells. We hypothesise that the development of a persistent infection within islet beta-cells might be critical to the initiation of islet cell autoimmunity and we are examining the molecular events that could underpin the development of such a response.
-

Hervé Perron & Jainy Thomas
University of Lyon - Geneuro
Identification of HERV-W copies and insertion sites associated with T1D in human genomesT1D etiology is not clearly understood, but it lies at crossroads between genetic predisposition and environmental factors. Research on the human genome has so far identified numerous candidate sequences as genetic risk factors, most notably within the HLA (Human Leucocyte Antigen) region. However, an important part of the human genome has been neglected in the sequencing studies conducted so far, mostly including the so-called “mobile genetic elements”, which represent about half of the human genome. HERV (Human Endogenous Retroviruses) belong to this group, as retro-transposable elements. Moreover, the envelope protein from HERV-W family (HERV-W-Env) has recently been associated with T1D.
Our goal is to identify T1D genetic risk factors belonging to mobile genetic elements, with an emphasis on HERV and HERV-W-env sequences. For this purpose, we propose to use novel sequencing technologies, which have not been previously used on the genome of T1D patients. These sequencing methods enable to detect genomic regions with repetitive or homologous sequences and to identify somatic DNA recombinations or rearrangements in affected tissue.
HERV-W-env sequences and most of HERV sequences have so far been ignored by traditional sequencing methods. Sequencing technologies used in this project may allow us to identify individuals harboring HERV-W- env pathogenic DNA copies and to develop targeted therapies neutralizing the protein before the clinical onset of T1D.
-

Edward Phelps & Todd Brusko
University of Florida/Immunocore
Funded by: Immunocore Ltd.
ImmTAAI technology and its potential use to treat autoimmunityWe propose an industry collaboration between investigators in the University of Florida Diabetes Institute and an industry partner, Immunocore Ltd.
Immunocore utilizes its T cell receptor (TCR) technology to generate tissue-targeted immune suppressant bispecifics called ImmTAAI (Immune modulating monoclonal TCR Against Autoimmune Disease) molecules. ImmTAAIs consist of a high affinity TCR-based targeting domain specific for a tissue-restricted peptide-HLA complex (pHLA), fused to an immune suppressive effector that inhibits autoreactive T cells (Fig 1). ImmTAAIs are designed to bind to target cells under autoimmune attack (e.g. pancreatic beta cells) and protect them from T cell driven destruction while not affecting the immune system elsewhere in the human body.
Here, we propose to profile the ImmTAAIs in a disease-relevant system, a human pancreas tissue slice / T cell coculture that reproduces the autoreactive T cell attack towards beta cells in a meaningful environment. We will first test the specificity of a pre-proinsulin (PPI) ImmTAAI to bind beta cells in the complex native pancreatic environment using live tissue slices.
-
Edward Phelps & Clayton Mathews
University of Florida
Funded by: NIH/NIDDK
Live human pancreas tissue slices as a platform for investigating the immunopathological processes of type 1 diabetesType 1 diabetes (T1D) is characterized by the loss of insulin producing β cells precipitated by infiltration of the pancreas by autoimmune T lymphocytes. We have shown that there is a parallel loss of function in the mass of β cells still remaining when T1D is diagnosed. This project aims to better understand how and why the remaining β cells stop working in people with T1D. Our main goal is to characterize the changes in the islet metabolism that cause these β cells to fail and the spatial and temporal relationship between these changes and the infiltration by T cells. In the first part of the study, we will look at how β cells lose their ability to produce insulin in response to glucose. We believe this is caused by early problems in how β cells sense sugar levels and transduce the signal to secrete the appropriate amount of insulin. Using donated human pancreas tissue, we will track these changes at the molecular level using advanced techniques like spatial metabolomics and test whether we can restore normal function in the β cells. The second part of the study will explore how the immune system interacts with β cells during disease development. We will compare β cell function in islets with different levels of immune cell activity using high thoughput encoding of islet function. Even when immune cells aren’t actively attacking, they may leave behind signs of previous damage. We will relate islet function with markers of active and past immune activity in the tissue samples. Finally, we will attempt to build a model of T1D in slices by co-culturing slices from non-diabetic donors with T cells transduced with diabetogenic T cell receptors. Together, these studies will provide a clearer picture of how and when β cells begin to fail in T1D. Our expert team combines strengths in imaging, metabolism, and immunology to work toward new ways to detect, prevent, or treat this disease.
-

Matthew Poy
Johns Hopkins All Children’s Hospital
Functional Role of CADM1 during Type 1 Diabetes PathogenesisType 1 diabetes (T1D) is characterized by the hyperglycemia resulting from the autoimmune destruction of the insulin-expressing β-cells of the pancreas. As the prevalence of the disease continues to increase worldwide, it remains imperative to identify therapeutic strategies that can (1) preserve pancreatic beta-cell mass and function and (2) prevent the immune response that initiates the pathogenesis of T1D. Cell adhesion molecule 1 (referred to as human CADM1 and mouse Cadm1, and also known as SynCAM1, TSLC1, Igsf4a, and Necl2) is an immunoglobulin-domain-containing membrane protein shown to mediate homo-and heterophilic cell-to-cell contact with other Cadm family members. We previously performed Cadm1 loss of function studies in β-cell lines and transgenic mice and established endogenous Cadm1 as a negative regulator of exocytosis and proliferation in pancreatic β-cells. It has also been shown that Cadm1+ cells will bind CD8+ T-cells via the receptor Class 1-restricted T-Cell-associated Molecule (Crtam) present in the immune cells; however to date, Cadm1-mediated cellular interactions have not been studied in the context of T1D. Here we hypothesize that CADM1 expression within the human pancreatic islet mediates cell-to-cell contact with infiltrating immune cells via the interaction of CADM1 present in alpha and beta cells with CRTAM on CD8+ T cells.The goal of this work is to perform immunohistochemical analysis to visualize CADM1isletexpression (in both alpha and beta cells) with markers of immune cell populations (i.e. CD8) within the islet infiltrate. This work seeks to: (1)identify CADM1 as a mediator of cell contact between specific immune cell populations and islet endocrine cells (both alpha and beta)and (2) validate the co-localization of CADM1 in the alpha and beta cell and CRTAM in CD8+ T-cells or NK immune cell populations. Completion of the proposed studies will determine whether CADM1 may be a potential therapeutic target for preserving beta cell mass and function and for attenuating the inflammatory response during the pathogenesis of T1D.
-
Matthew Poy
Johns Hopkins All Children's Hospital
Funded by: Helmsley Charitable Trust
Investigation of Myeloid CADM1 during T1DType 1 diabetes (T1D) is characterized by the hyperglycemia resulting from the autoimmune destruction of the insulin – expressing β – cells of the pancreas. As the prevalence of the disease continues to increase worldwide, it remains imperative to identify therapeutic strategies that can (1) preserve pancreatic beta – cell mass and function and (2) prevent the immune response that initiates the pathogenesis of T1D. Cell adhesion molecule 1 (referred to as human CADM1 and mouse Cadm1, and also known as SynCAM1, TSLC1, and Necl2) is an immunoglobulin – domain – containing membrane protein shown to mediate homo – and heterophilic cell – to – cell contact with other C ADM family memb ers. It has also been shown that C ADM 1+ cells will bind CD8+ T – cells via the receptor Class 1 – restricted T – Cell – associated Molecule (C RTAM ) present in the immune cells; and we recently showed the number of CADM1 – expressing myeloid cells are increased within the human pancreatic islet during human T1D. These results suggest CADM1 – expressing populations may contribute to the recruitment and activation of CD8+ T cells that leads to destruction of beta – cells . Preliminary results show C ADM 1- expressing myeloid ce lls in islets of NOD mice are also enriched for expression of Toll – like receptor 7 (TLR7) and DC – SIGN, two established activators of innate immune cell populations shown to bind a broad range of pathogens . The goal of this work is to perform (1) multiplex immunohistochemical analysis to quantify CADM1+ myeloid cells that express TLR7 and DC – SIGN within islets of auto – antibody positive and T1D subjects and (2) spatial transcriptomic analysis to identify additional co – factors of CADM1 – expressing cells during T1D . This work seeks to establish that increased numbers of CADM1 – expressing myeloid cells are in proximity to CD8+ T cells in islets of aAb+ and T1D human subjects. Completion of the proposed studies may establish CADM1+ TLR7+ DC – SIGN+ cells as potential therapeutic targets for preserving beta cell mass and function and for attenuating the inflammatory response during the pathogenesis of T1D. -

Alberto Pugliese
Diabetes Research Institute, University of Miami
The nPOD-Virus GroupnPOD has now assembled a “cloud” of investigators with diverse expertise, who are interested in collaborative studies, and is ready to take the nPOD research model to a higher level: the nPOD working groups. These groups are intended to collectively tackle key questions in diabetes research.
The nPOD virus working group (nPOD-V) represents a self-assembled a collaborative effort with the goal of investigating the role of viruses in type 1 diabetes through the study of nPOD samples. An innovative concept implemented by the group is “real-time” data sharing, to help coordinate studies and inform strategy adjustments. This approach should accelerate the rate of discovery and maximize the potential for robust results. The premise underlying this collaboration, based on the nPOD research model and the concept of data sharing and collaboration promoted by the nPOD leadership, has been fully embraced by the nPOD-V investigators; we believe that this in itself represent a major step-forward and innovation in the study of complex questions in human disease.
Many studies have linked enterovirus infection to islet autoimmunity and diabetes. Yet, many questions remain about which virus serotypes are linked to T1D, what type of infection they cause, and how this may contribute to the autoimmune process leading to diabetes. Earlier studies from individual laboratories suffered from limitations with samples and reagents, and did not have access to the latest methodological advances. This collaborative effort recognizes those limitations, and addresses them with an integrated and multidisciplinary approach that includes many innovative and powerful technologies that have not been used before for studying viruses in type 1 diabetes. The availability of shared tissues from the same patients and their coordinated analysis provide unprecedented opportunity that investigations can be exhaustive and the most informative.
The precise identification of enterovirus serotypes associated with T1D and a better molecular characterization of the virus host interaction and relation to pancreas pathology could lead to the identification of novel, T1D-specific, biomarkers of infection and potentially novel therapeutic targets. If a virus plays a role in islet autoimmunity, a vaccine or drugs that target viral responses could perhaps become important therapeutic avenues for preventing the triggering of autoimmunity or its progression, reducing patient burden and potential exposure to immunosuppressive or immunomodulatory treatments which can be associated with significant side effects and long term concerns, especially when children are concerned.
-
Nadav Sharon
Technion - Israel Institute of Technology
Funded by: EFSD
Identification of protective niches in T1D pancreataType 1 diabetes (T1D) is caused by an autoimmune attack mediated by cytotoxic T cells. However as the attack unfolds, many other cell types participate–and some play an anti-inflammatory, regulatory, role. Interestingly, in the early stages of the disease, attacked islets often reside next to non-attacked islets. We hypothesized that the “tranquil” islets harbor elements which protect against the immune attack, and so ventured to compare attacked and non-attacked islets in pancreata at the pre-symptomatic stage of the disease. In this framework, we characterize the different cell populations which reside in either the attacked or non-attacked islet niche. Specifically, we focus on a population of macrophages with anti-inflammatory characteristics which reside exclusively in non-attacked islets. We now study the potential role of these cells in attenuating attack progression, and look for ways to activate them as means to delay–or even prevent–T1D.
-
John Todd, Rachael Bashford-Rogers & Sarah Richardson
University of Oxford
Funded by: Breakthrough T1D and Wellcome Trust
Characterising the organisation of the gut associated lymphoid tissue in type 1 diabetes and tracking T and B cell receptor clonotype / isotype usage and islet antigen-specific lymphocytes across tissues in the nPOD resource.The intestinal microbiome plays an essential role in host health, where microbial communities have immune-modulating activity and impact on regulatory immune cell compartmentalisation in the gut associated lymphoid tissues. Increasing evidence points to a dysfunctional relationship between the microbiome and host tissues in individuals who develop type 1 diabetes (T1D). Alterations in gut microbial composition and diversity have been observed in cohorts of individuals who go on to develop diabetes and the amount of antibody binding to common microbes is altered in diabetics, controlled in part by T1D risk HLA haplotypes. Antibodies play a key role in the composition and healthy function of the gut microbiota. Development of the most aggressive form of T1D, with the near complete destruction of insulin producing beta cells in children diagnosed under the age of 7, is associated with an increased number and proportion of B lymphocytes infiltrating the pancreas. Using samples from tissues across affected and systemic tissues in T1D (duodenum, spleen, pancreatic lymph node and pancreas tissues) from nPOD, we will test the hypothesis that perturbations in microbial populations and intestinal barrier function promote cross-reactivity to pancreatic beta-cell antigens thus driving development of T1D in children. We will apply a sequencing approach to deeply characterise the mucosal-associated microbial community in intestinal tissues derived from control, autoantibody positive and T1D donors. In parallel, in our approved study, we will test whether T and B cells present in the gut are related to those found in the spleen, pancreatic lymph nodes and the pancreas using a sequencing-based approach to identify cells with a shared ancestry. We will trace related clones across sites through the B/T cell receptor sequences and determine their binding characteristics using cloning and expression approaches to test for microbial and pancreatic beta-cell specificity.
-
Clive Wasserfall & Harry Nick
University of Florida
Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein and the Autoantigens GAD, IA-2 and ZnT8 in Type 1 Diabetes PancreataThe canonical notion that type 1 diabetes (T1D) results following a complete destruction of β cells has recently been questioned as small amounts of C-peptide are detectable in patients with long-standing disease. Previously, we analyzed protein and gene expression levels for proinsulin, insulin, C-peptide, and islet amyloid polypeptide within pancreatic tissues from T1D, autoantibody positive (Ab+), and control organs. Insulin and C-peptide levels were low to undetectable in extracts from the T1D cohort; however, proinsulin and INS mRNA were detected in the majority of T1D pancreata. Interestingly, heterogeneous nuclear RNA (hnRNA) for insulin and INS-IGF2, both originating from the INS promoter, were essentially undetectable in T1D pancreata, arguing for a silent INS promoter. Expression of PCSK1, a convertase responsible for proinsulin processing, was reduced in T1D pancreata, supportive of persistent proinsulin. These data implicate the existence of β cells enriched for inefficient insulin/C-peptide production in T1D patients, potentially less susceptible to autoimmune destruction. Moving forward, we propose to expand these studies to address the molecular mechanisms, including those of genetics (at a single cell to whole pancreas level) that contribute to novel aspects of T1D development, including the stress responses, protein misfolding, and mitochondrial dysfunction. Follow up experiments will determine the relationship of autoantigens to autoantibody titer. It is well known that autoantibodies wane after the onset of type 1 diabetes. Herein we are investigating the rate of loss of autoantibodies post onset with loss of these target proteins in the beta cells.
Archived Projects
-
Reza Abdi & Marwan Mounayar
Brigham and Women’s Hospital
Funded by: Breakthrough T1D
Intrinsic immunoregulatory defect of bone marrow mesenchymal stem cells (BM-MSC) from T1D patientsType 1 diabetes (T1D) represents a major public health burden demanding innovative treatment strategies. Mesenchymal stem cells (MSC) have profound immunomodulatory effects. Numerous clinical trials have focused on the immunomodulatory capacity of MSC to treat various immune-mediated diseases. We were amongst the first to show the protective effects of allogeneic MSC in autoimmune diabetes in NOD mice. More studied need to be carried out in particular on human MSC to unravel the mechanisms of immunomodulation of MSC to help in the design of MSC therapy in T1D. We are fortunate to collaborate with nPOD to receive samples from T1D patients to isolate and grown MSC for our studies to examine their immunomodulatory capacity. Data to be generated using these samples are crucial in better understanding as how MSC suppress autoimmunity which would support the bases for These data provide important preclinical data supporting the basis for further development of MSC-based therapies for T1D and potentially, for other autoimmune disorders.
-

Vitaly Ablamunits, Kevan Herold & Jasmin Lebastchi
Yale University
Funded by: Yale DERC
Comparison of total pancreatic insulin content and peripheral markers of beta cell function and desctructionIn both Type 1 and Type 2 diabetes, there is a gradual loss of pacreatic beta cells with time. The degree of beta cell destruction may be indirectly measured by the appearance of islet-specific components in circulation. For instance, insulin mRNA is increased in circulation in T1D patients before clinical loss of function of transplanted islets. We have recently demonstrated, that hypomethylated insulin gene characteristic for beta cells leaks into circulation during beta cell death and can predict hyperglycemia in both STZ-induced and spontaneous (NOD) mouse models. In pre-diabetic NOD mice, hypomethylated proinsulin DNA in circulation negatively correlated with total pancreatic insulin content measured in acid-ethanol extracts. However, in a natural history of prediabetes, the destruction may be slow at the very beginning due to low numbers of effector T cells, or be slow immediately before clinical precipitation of diabetes, because most of the target beta cell mass has been lost already. Thus the same level of destruction may have different prognostic value. The only accurate way to interpret the level of destruction would be to know residual beta cell mass. For that, in clinical setting, surrogate markets have been traditionally used, such as C-peptide, or, more recently, beta cell mass have been visualized by positron emission tomography. However, in humans, this data has never been directly correlated to total pancreatic insulin content. We hypothesize that circulating C-peptide (amylin, proinsulin) levels strongly correlate with total pancreatic insulin content, and that a ratio between the biomarker of beta cell death and C-peptide determines prognosis.
-

Domenico Accili & Chutima Talchai
Columbia University
Funded by: NIH
Beta cell dedifferentiation in type 2 diabetesDiabetes is associated with β-cell dysfunction. But it remains unclear whether the latter results from reduced β-cell number or function. Purpose of this study is to investigate the hypothesis that β-cell failure is the result of β-cell dedifferentiation, rather than apoptosis or degranutaion. Transcription factor FoxO1 integrates β-cell proliferation with adaptive β-cell function. The rationale for the proposed project arises from the observation that mice lacking FoxO1 in β-cells develop hyperglycemia with reduced β-cell mass following physiologic stress, such as multiparity and aging. Surprisingly, lineage-tracing experiments demonstrate that loss of β-cell mass is due to β-cell dedifferentiation, not death. Dedifferentiated β-cells revert into progenitor-like cells expressing Neurogenin3, Hdac1,and L-Myc. They also partly convert into non-β-cells, resulting in hyperglucagonemia. Strikingly, we identify the same sequence of events as a feature of different models of murine diabetes. We intend to investigate whether humans with type 1 and 2 diabetes develop the same abnormalities observed in Foxo1 β-cell-specific knockouts.
-

Taro Akiyama
Merck
Confirmation of target gene expression in T1D and T2D pancreasCharacterization of expression of novel anti-diabetes targets in pancreas of Type 1 and Type 2 diabetics.
-

Mark Stuart Anderson
University of California, San Francisco
Funded by: Breakthrough T1D
Identification of novel Tissue-Specific Antigens expressed by human Extrathymic Aire-Expressing Cells, and determination of their potential contribution to the prevention of type 1 diabetesAire+ cells were readily identified by immuno-staining in both PN and nPLN sections. Like in mouse, human eTACs were relatively rare but localized outside the B-cell follicles and stained with the hallmark “nuclear speckling” pattern. Furthermore, the eTACs were ubiquitously positive for MHC class II but lacked high expression of CD11c, calling into question their identification as a known resident DC population. Quantitative PCR confirmed the expression of Aire in whole PLN and in sorted CD45+, MHC class II+ populations from fresh human spleen. No difference was found in the relative expression of Aire between T1D, no diabetes, and pre-T1D PLN specimens. However, the expression of ladinin-1, a putative Aire-regulated TSA in mouse eTACS, was positively correlated with Aire expression, suggesting that it might be an Aire-regulated TSA in humans as well.
Overall, our data confirm that Aire is indeed expressed outside the thymus in humans and that human eTACs appear analogous to the more fully characterized murine eTACs. Their potential for promoting peripheral tolerance in a therapeutic setting remains an exciting possibility.
-

Justin Annes
Stanford University
Funded by: NIDDK
Beta cell replicationRecent decades have seen an incredible growth in the prevalence of diabetes. While it is important to pursue multiple therapeutic strategies, harnessing the regenerative capacity of islet β-cells to increase an individual’s insulin secretion capacity is among the promising approaches.
Recently, discovery-oriented unbiased chemical screening led to the identification of the metabolic enzyme adenosine kinase (ADK) as a regulator of rodent and porcine β-cell replication. Importantly, small molecule ADK-inhibitors selectively stimulate replication of β-cells, but not alpha-cells, in vitro and in vivo. The identification of ADK as a regulator of β-cell regeneration has raised several important questions.
Does long-term inhibition of ADK in vivo promote β-cell mass expansion and protect against developing diabetes? How does a broadly expressed metabolic enzyme selectively control the growth of a specific cell-type?
Is human β-cell growth controlled by ADK? Using the nPOD resources, we are investigating the expression of ADK in normal and diabetic pancreatic tissues. We are interested in weather this molecule is expressed in human β-cells and whether its expression changes with diseased states.
-
Mark Atkinson, Irina Kusmartseva, Clive Wasserfall & Todd Golde
University of Florida
Microtubule-associated protein TAU and the Islets of LangerhansTau is one of the most important known molecules for microtubular dynamic and trafficking within the cells. In neurogenerative disorders such as Alzheimer disease, the tau becomes hyperphosporylated and form insoluble intracellular protein aggregates leading to upregulation of unfolded protein response. At the same time, patients with Alzheimer’s disease show impaired glucose metabolism. Moreover, both insulin-dependent and non-insulin-dependent diabetes lead to cognitive deficits in diabetes patients. Recent studies demonstrated the presence of TAU and its hyperphosphorylated pathogenic forms in islets of Langerhans in Type 2 diabetes patients,suggesting an potential role of TAU in pancreatic endocrine cell function and insulin secretion. Thus, this project seeks to elucidate presence and distribution of total and hyperphosphorylated TAU protein in endocrine cells of pancreatic islets and establish whether accumulation of hyperphosphorylated TAU is associated with aberrant islet unfolded protein response in type 1 and type 2 diabetes.
-
Mark Atkinson & Anil Bhushan
University of Florida & University of California, San Francisco
Beta Cell Senescence and T1DType 1 diabetes (T1D) results from an autoimmune disorder characterized by T-cell-mediated destruction of pancreatic beta cells. While the role of auto-reactive T cells in targeting beta cells has been well defined, the pathological mechanisms operating in beta cells remains poorly understood. To investigate beta cell stress responses during disease progression but before diabetes onset, we performed single-cell RNA-seq (scRNA-seq) on islet cells from 8 week and 14 nonobese diabetic(NOD) mice. This analysis revealed two transcriptionally-distinct sub-populations of beta cells in the 14 week beta cells, which had hallmarks of stress response senescence and the senescence-associated secretory phenotype (SASP). The SASP consists of proinflammatory cytokines, chemokines, extracellular matrix factors, metalloproteases and growth factors secreted from senescent cells that regulate the tissue microenvironment and provide signals for immune surveillance and senescent cell clearance. As the SASP has been previously implicated in the pathogenesis of a variety of age-related diseases, we hypothesized that accumulation of SASP beta cells could be a contributing factor to disease progression in T1D. Consistent with our hypothesis, further studies revealed that targeted elimination of SASP beta cells with small molecule inhibitors prevented T1D in the NOD mouse model.To determine whether beta cells undergo senescence and SASP during the natural history of T1D in humans, we have already performed two studies on a cohort of nPOD specimens from similarly age nondiabetic, autoantibody-positive and T1D donors. Xgal staining of cryosections was used to detect the activity of senescence-associated βgalactosidase (SA-βgal), a sensitive and widely used biomarker for senescent cells in vivo. This analysis showed dramatically elevated SA-βgal activity in the islets of T1D donors as compared with nondiabetic and autoantibody-positive donors, consistent with a higher number of senescent beta cells. We also performed immunohistochemistry for senescence (CDKN1A, CDKN2A, Ser139 phospho-H2A.X) and SASP (IL-6, IL-8, MMP9, SERPINE1) markers. While nondiabetic donors generally had no expression of these markers in beta cells, autoantibody-positive and T1D donors had significantly increased levels. These data extend and build upon our findings in the NOD mouse model and suggest involvement of beta cell SASP inthe progression of human T1D. In order to test the hypothesis that beta cell SASP plays a role in T1D pathogenesis in humans, as well as to investigate whether hallmarks of SASP could provide novel biomarkers for disease progression, the following experiments requiring nPOD specimens are planned:1) Evaluate exosome RNA biomarkers in nPOD donor serum/plasma. We will evaluate candidate exosome small noncoding RNA biomarkers identified from small RNA-seq analysis of SASP islet exosome RNA human serum from nondiabetic, autoantibody-positive and T1D donors; and 2) Evaluate therapy for targeted elimination of human SASP beta cells using nPOD donor islets. Using our current small molecule therapy for clearance of SASP beta cells in NOD mice and islets, we will perform similar studies on nPOD donor islets in culture to provide proof-of-concept of this therapy for ablating human SASP beta cells.
-

William Bachovchin
Tufts University
Funded by: NIH
Fibroblast Activation Protein in Type 1 DiabetesFibroblast activation protein is an extracellular membrane bound and soluble protease that is commonly used as a marker of activated fibroblasts. Expression of FAP in healthy adult tissues is limited, but up-regulation is observed in a number of inflammatory and tissue remodeling processes, including wound healing, liver fibrosis and epithelial cancer, where it is thought to participate in remodeling of the extracellular matrix via its protease activity.
We hypothesized that FAP would also be up-regulated in the pancreatic remodeling and inflammatory process associated with type I diabetes. FAP has the potential to participate in this disease process in a number of ways. First, it has been suggested that immune cells cannot infiltrate pancreatic islets unless the membranous capsule is destabilized in some way. As a protease with collagenase activity, FAP may participate in degradation of the basement membrane allowing immune cell infiltration into the islets. Second, FAP may cleave hormones involved in glucose metabolism. The FAP KO mouse has been reported to have improved glucose tolerance, particularly after high fat feeding, suggesting as yet undiscovered substrates may exist for FAP. Preliminary work in our lab has identified FAP cleavage of a number of metabolically active hormones in vitro and FAP consensus sequences exist in many soluble factors including the hormone betatrophin. Thus FAP may have substrates of consequence related to beta cell regeneration or glucose control.
The goal of this project is to quantify FAP activity in the pancreas and serum of type 1 and type 2 diabetics compared to controls. This study will also be, to our knowledge, the first to examine FAP activity in the human pancreas.
-

Manuela Battaglia
San Raffaele Scientific Institute, Italy
Funded by: Breakthrough T1D
Dissecting the effector/regulatory compartments in the target tissues of T1DAn altered balance between pathogenic and regulatory pathways in autoimmunity has been hypothesized and at times demonstrated in peripheral blood of patients with type 1 diabetes. However, our knowledge in humans at the sites of the autoimmune attack (i.e., the pancreas) is scanty. Studies generated in our lab. demonstrate that FOXP3+ T regulatory (Treg) cells, identified based on their epigenetic imprinting, are present within the pancreatic lymph-nodes of patients with type 1 diabetes but they lack a suppressive activity towards both polyclonal and insulin-specific effector responses. We have found that there is an important discrepancy in Treg cells of T1D patients between their epigenetic imprinting, FOXP3 expression and regulatory function when these cells reside in the draining lymph nodes but not when they circulate in the blood.
Given the crucial role that microRNA (miRNA) genes have demonstrated in modulating immune responses, it is possible that dysregulation of miRNA-expression contributes to the pathogenesis of autoimmune diseases, type 1 diabetes included, as recently demonstrated in other autoimmune diseases (e.g. multiple sclerosis).
Objectives of our work are: (i) to characterize the miRNA expression profile in Treg cells residing in different tissues of the same donor (healthy and with type 1 diabetes), and (ii) to understand whether Treg cells residing in the lymph-nodes of T1D patients are post-transcriptionally regulated in a different way than those isolated from healthy donors. Results from this study may be instrumental for our comprehension of type 1 diabetes and for the development of novel therapeutics to target miRNAs to modulate immune responses.
-
Manuela Battaglia
San Raffaele Hospital
Funded by: Breakthrough T1D
Innate immunity and diabetesAlthough T1D is considered to be mainly a T-cell mediated and a beta-cell specific disease we have generated over the years the hypothesis (and some supporting data) that innate immunity is also key in disease initiation and that T1D might be a disease of the whole pancreas that manifests in autoimmunity to beta cells.
Our published data indicate that neutrophils infiltrate the pancreas of patients with T1D [1] and our still unpublished results suggest that peripheral markers of Neutrophil Extracellular Traps (NETs: a networks of extracellular fibers composed of DNA extruded from neutrophils, which can bind pathogens but can also lead to tissue damage and dangerous exposure of self antigens) are found increased patients with T1D. In addition, we have evidence that neutrophils infiltrate the whole pancreas and not only the islets [1] (a phenomenon also reported for CD8+T cells [2]). Neutrophils can also exert interesting functions when invade tissues (e.g.,production of neutrophil extracellular traps -NETs, interaction with other innate immune cells, and source of post-translationally modified self antigens) –functions that overall we call the “innate network”.
Thus, the objective of this project is to test the hypothesis that neutrophils and NETs are increased in AAb pos and T1D nPOD donors. We propose to use the POD cohort to identify innate immune cells and their markers of activity. These important samples will allow us to test our hypothesis and –if confirmed–to design new therapeutic approaches.
ADDENDUM: Although T1D is considered to be mainly a T-cell mediated and a beta-cell specific disease we have generated over the years supporting data that innate immunity is also key in disease initiation and that T1D might be a disease of the whole pancreas that manifests in autoimmunity to beta cells.
Our published data indicate that neutrophils infiltrate the pancreas of patients with T1D [1] and peripheral markers of Neutrophil Extracellular Traps (NETs: a networks of extracellular fibers composed of DNA extruded from neutrophils, which can bind pathogens but can also lead to tissue damage and dangerous exposure of self antigens) are found increased in patients with T1D. In addition, we have evidence that neutrophils infiltrate the whole pancreas and not only the islets(a phenomenon also reported for CD8+T cells2). Neutrophils can also exert interesting functions when they invade tissues (e.g.,production of neutrophil extracellular traps -NETs, interaction with other innate immune cells, and source of post-translationally modified self antigens) –functions that overall we call the “innate network”.
We recently generated new data3demonstrating that: (i) purified circulating neutrophils have a specific gene-signature in pre-symptomatic subjects and in patients with T1D. This signature is already present at very early stages of the disease (i.e., in autoAb negative subjects); (ii) NETting neutrophils (cells that have protruded Neutrophil Extracellular Traps) are present in the pancreas of subjects with recent onset T1D and autoAb positive donors but not in non-diabetic controls.
Thus, the objective of this “addendum” is to isolate pancreas infiltrating neutrophils for transcriptomic characterization and validate the peripheral correlate through the DepArray™ technology.We propose to use the nPOD cohort to delineate the transcriptomic signature of neutrophils attracted into the pancreas of pre-symptomatic and symptomatic T1D subjects that would differ from that of non-diabetic donors.
These important samples will allow us to test our hypothesis and –if confirmed –to design new therapeutic approaches.
-
Graeme Bell & Manami Hara
University of Chicago
Beta-cell Death and SurvivalType 1 diabetes (T1D) is a progressive immune-mediated disease that is preceded by an asymptomatic preclinical period of highly variable duration. A better understanding of beta cell and islet function or dysfunction during this asymptomatic period and the nature of the “dialog” between islet endocrine cells and the immune system could lead to new therapeutic strategies to arrest the progression to autoimmunity. Here we propose to develop a framework for understanding the underlying biology of T1D by integrating histological and genomic (RNA-seq) analyses of the human pancreas, pancreatic islets and islet endocrine cells. We anticipate that these studies will lead to a new understanding of the pathophysiology of T1D and point to biomarkers of human beta-cell stress, death and survival.
-

Jeffrey Bluestone & Armando Villalta
University of California, San Francisco
Vascular endothelial growth factor receptor signaling regulates the pathogenesis of type 1 diabetes1. To test the hypothesis that vascular endothelial cells in the pancreas of recent-onset diabetic patients have increased expression of VEGF receptors (VEGF-R).
2. To examine the expression of VEGF or VEGF-R on islet-invading CD4+ T lymphocytes.
3. To examine the utility of TCR repertoire between different T cell subsets as a marker of T1D disease initiation and progression by high throughput sequencing. -

Bernhard Boehm & Prabhakar Shyam
Nanyang Technological University
Funded by: A*STAR Singapore and LKCMedicine
Validation of blood – derived single cell gene expression signature in peri – pancreatic lymph nodes of Type 1 Diabetes subjectsType 1 diabetes (T1D) is an organ specific autoimmune disease resulting in an immune-mediated destruction of insulin-secreting beta cells. The etiology of T1Dis puzzled by a multifaceted interaction of environmental, genetic and epigenetic factors leading to variousendotypesof the disease. Fine mapping of T1D-associated genetic variants with cell-specific epigenetic markers or expression quantitative traits (eQTLs) indicates that the disease riskfactors most probably exert their function by changing the transcriptome profile of immune cells in a cell specific manner. This explains the heterogeneous nature of T1D in terms of both clinical representation and underlying pathogenic pathways. Therefore, measuring the cell-specific transcriptome profile and composition of peripheral immune cells is considered as the key to identify the exact cellular and molecular pathogenic mechanisms of the diseaseprogressand develop novelimmunesignatures for monitoring disease progression.In spite ofextensive research into T1D-associated immune signature, there is still no consensus about T1D-associated immune cell typesand their pathogenic signature.By taking advantage of the unbiased single cell RNA sequencing technology, we profiled transcriptome landscape of peripheral blood mononuclear cells (PBMCs) derived from 40 European T1D subjects and 35 healthy controls matched for age, gender and HLA alleles. We detectedsignificant alteration in relative frequency of several immune cell types(particularly subtypes of CD4+ T, CD8+T, and regulatory Tcells) inT1D subjects compared with healthy controls. Interestingly, we found that the aberrant transcriptome profile of these cell types issignificantly enriched in immune pathwaysassociated with enhanced antigen priming capacity(DAP12 signaling pathway)and transendothelial migratory potential(Rho GTPase activate ROCK pathway) of T cells. Since it is well known that peri-pancreatic lymph nodesare essential for the priming of auto-reactive T cellsin T1D, we hypothesized that the observed signatures in circulating blood cells root from the peri-pancreatic lymph nodes wherein enhanced antigen priming and trans-endothelial migration potential of activated cells lead to the pathogenesis of the disease. To test our hypothesis, the currentproposal aims tocharacterizeimmune subtype proportionand transcriptomic profilein the peri-pancreatic lymph nodes ofT1D cases and controls(aim 1), identify aberrantcell-specific genes/pathways (aim2) and cross-validate lymph node signature using single cell analysis of PBMCs from the same individuals as well as our single cellPBMC datasetfrom the European cohort(aim3). We envisage thatthe current research projectwill work as an essential step for the development of a blood-basedblueprint associated with pathogenesis and progression of the disease.
-

Marika Bogdani
Benaroya Research Institute at Virginia Mason
Funded by: Breakthrough T1D
Extracellular matrix involvement in type 1 diabetes pancreatic islet destructionThe overall purpose of our research is to understand how changes in extracellular matrix affect insulitis initiation and progression and beta cell destruction in diabetes.
Our research focuses on a specific extracellular matrix molecule, hyaluronan, a long chain glycosaminoglycan distributed widely throughout different tissues. Hyaluronan is highly abundant in inflamed tissues and its synthesis is responsible for many of the physiologic changes associated with inflammation, including edema, vascular permeability changes, and leukocyte egress and activation at sites of injury, as well as the maturation of dendritic cells, antigen presentation, and the function and number of regulatory T cells. Hyaluronan exerts these effects through interactions and formation of macromolecular complexes with hyaluronan-binding molecules called hyaladherins, such as inter-α-inhibitor, versican, and tumor necrosis factor-stimulated gene-6.
We have made the novel observations that hyaluronan and hyaladherins increase in pancreatic islets of younger donors and accumulate in regions of lymphocytic infiltrates in T1D, and that both the amount and distribution of these specific extracellular matrix molecules vary with time since diabetes onset. We have also shown that hyaluronan and hyaladherins are deposited in the follicular germinal centers and T-cell areas, regions of immune cell activation, in both human pancreatic lymph nodes and spleen in type 1 diabetes.
We are investigating (1) the regulation of hyaluronan accumulation in pancreatic islets and lymphoid tissue in T1D, (2) the functional significance of hyaluronan – hyaladherin complexes in initiation and progression of insulitis, (3) the effect of altered hyaluronan synthesis and hyaluronan interactions with hyaladherins on diabetes development, (4) the role of hyaluronan – hyaladherin matrices in modulation of immune cell interactions and their adhesive and migratory properties, and in promoting T-cell activation and proliferation.
Our novel findings were obtained from study of human samples provided by nPOD. Analysis of those tissues also forms the basis of the work in progress on changes in hyaluronan and hyaladherin matrices in human tissues in diabetes.
-

Peter Butler, Alexandra Butler & Mark Atkinson
University of California, Los Angeles
Pancreatic duct glands in type 1 diabetesThe initial overall question to be tested by the collaborative of the Peter Butler laboratory at UCLA and the Mark Atkinson laboratory in Florida was to explore the possibility that the recently described pancreatic duct gland (PDG) compartment of the pancreas might serve as the elusive pancreatic stem cell niche, potentially offering an avenue to promote beta cell regeneration. Both laboratories had previously noted the presence of pancreatic beta cells in long standing type 1 diabetes mellitus (T1DM) and had been interested in the possibility that these were derived from beta cell regeneration rather than simply reflecting beta cell survivors.
The initial studies focused on quantification of the PDG compartment in T1DM with comparison to type 2 diabetes mellitus (T2DM) and non-diabetic controls. Since the primary interest in the PDGs was as a potential source of endocrine cells, we also examined the nPOD immunostained sections for insulin and glucagon to evaluate if the PDGs did indeed contain insulin or glucagon staining cells. During this process it was noted that there some of the nPOD sections had markedly expanded glucagon staining compared to normal controls. Further examination of the possible explanation of this led to the observation that the increased glucagon staining was most prominent in the group of individuals with T2DM treated with GLP-1 based therapy prior to their demise.
Following communication between the collaborating laboratories in Florida and Los Angeles, it became apparent that we had a responsibility to further delineate the unexpected differences between the pancreases of individuals with T2DM treated by GLP-1 based therapy and other therapies. The need for this was further heightened by the finding by the University of Florida group of neuroendocrine tumors expressing glucagon in GLP-1 treated T2DM patients. Further studies by an experienced pancreatic pathologist at UCLA raised concern that there was increased pancreatic dysplasia in the pancreases, and data from nPOD revealed increased overall pancreas replication (by Ki67) and expanded pancreas size. A question arose as to how best to make these observations known. It was decided that it would be preferable to include all the findings that seemed relevant to these observations in a peer-reviewed paper as well as to inform the domestic regulatory agency in charge of drug safety (i.e., the Food and Drug Administration – FDA). After months of rigorous peer review, the work saw publication in May of 2013 in the journal Diabetes. In addition, following notification, the FDA posted a Drug Safety Communication in March 2013 noting that a group studying pancreatic pathology had suggested an increased risk of pancreatitis and pre-cancerous cellular changes in patients with type 2 diabetes treated with incretin mimetics.; however, the FDA could not reach any new conclusions about safety risks of incretin mimetic drugs. A vigorous response was seen from interested pharmaceutical interests including broad based subpoenas to multiple individuals at UCLA. The current work in relation to the nPOD studies is now focused largely on repeating various aspects of the study (e.g., glucagon immunostaining originally preformed by nPOD at the University of Florida now being repeated at UCLA) to establish to what extent the criticism of the studies is valid (in which case the authors will acknowledge this) or if not, then respond with additional findings. Since the initial FDA statement, they have subsequently published a follow up finding together with EMA (European Medicines Agency) in February 2014 in the New England Journal of Medicine. These agencies concluded that, based on current data, a causal association between incretin-based drugs and pancreatitis or pancreatic cancer could not be drawn.
In this regard the University of Florida group is now preparing a publication for peer review supporting the notion that the pancreas mass is indeed increased in individuals treated with GLP-1 based therapy. Moreover, the expansion is most apparent in the head of the pancreas, the region most vulnerable to pancreatic dysplasia.
The ongoing UCLA based studies to re-evaluate the alpha cell hyperplasia in GLP-1 treated individuals previously reported from the nPOD immune staining, has agreed with the conclusion of the prior analysis, that there is alpha cell hyperplasia in the incretin treated cases.
Once the issues of criticism in regards the prior study have been addressed the plan is to revert to the originally intended focus, specifically on the PDG compartment as a potential source of endocrine cell formation in humans.
-

Alexandra Butler, Sangeeta Dhawan
University of California Los Angeles
Funded by: National Institutes of Health/NIDDK and Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
ORIGINAL: Growth and Development of the Endocrine Pancreas in Humans: Extrapolating Normal Physiology to the Regenerative Capacity of the Endocrine Compartment in Type 1 and Type 2 Diabetes; ADDENDUM: The Role of Neuropeptide Y (NPY) in Human Beta Cell Maturation and Diabetes PathogenesisORIGINAL: Chromogranin A is a global marker of endocrine cells. Most endocrine cells in the pancreas, in addition to Chromogranin A, also express other hormones, Insulin and Glucagon being most frequent, with Pancreatic Polypeptide, Somatostatin and Ghrelin in smaller numbers of endocrine cells. We have found that in adult human pancreas a small number of pancreatic endocrine cells (<1% of the total number) express Chromogranin A but not any of the other known pancreatic hormones; we have termed these cells Chromogranin Positive Hormone Negative [CPHN] cells.
We found that there is a very high percentage of CPHN cells in sections from human fetus and newborn pancreas, which suggests that these cells are a precursor of mature endocrine cells. We also found that the percentage of CPHN cells is higher in pancreas from both type 1 and type 2 diabetic subjects, suggesting that this may be an attempt at restoring the beta cells lost as a consequence of disease.
Our initial observations are that these cells occur mostly in small clusters of 1-3 cells rather than in established islets, suggesting that these cells may be newly formed endocrine cells. The source of these CPHN cells is of great interest, as they could potentially occur de novo (neogenesis), by transformation of existing cells (transdifferentiation) such as from cells in the ducts or exocrine pancreas, or by replication of existing endocrine cells.
Our goal is to characterize these cells by looking at the frequency of replication and apoptosis , and the transcription factors Nkx6.1 and Nkx2.2, in the CPHN cell population. Our findings will have significance with regard to the regenerative capacity of human endocrine cells in human pancreas.
ADDENDUM: Both type 1 and type 2 diabetes result from loss of functional beta-cell mass. Functional defects in evolving T1D and T2D are marked by the uncoupling of ambient glucose and insulin secretion, reminiscent of functionally immature beta cells in early pancreatic development. To develop successful beta-cell replacement for reversal of beta-cell dysfunction in diabetes, it is therefore critical to understand mechanisms that regulate beta cell maturity. Beta cells are formed during embryonic life, but acquire function only after birth, in a process called beta cell maturation. We have identified Neuropeptide Y (NPY) as a marker of functionally immature, neonatal beta cells. NPY is known to inhibit insulin secretion, and we show its co-localization to the insulin secretory granules in neonatal human beta cells. Our preliminary data show that NPY is down-regulated in adult beta cells, and re-expressed in beta cell in mouse models of type 2 diabetes. Thus, we propose to examine NPY in human beta cell maturation and diabetes pathogenesis.
-

John Cambier & Peter Gottlieb
University of Colorado, Denver
Tracking the loss of anergic B cells in pre-diabetic and new onset T1DHLA TestingInsulin-specific B lymphocytes play an important role in development of autoimmunity in Type 1 Diabetes (T1D). Since potentially offensive autoreactive cells are silenced by mechanisms of immune tolerance, participation of insulin-binding B cells (IBCs) in T1D must reflect escape from this silencing. Examining this question, we found that those IBCs bearing antigen receptors with high affinity for insulin occur only in the anergic BND (naive IgD(+)IgM(-) B cells [Duty, et.al. 2009 J Exp Med. 16: 139-51]) B cell compartment in peripheral blood of healthy subjects. Importantly, these cells are absent from this compartment in some “at risk” first degree relatives and all pre-diabetic and new-onset (<1yr) T1D patients. BND cells were found at normal levels in individuals diabetic for >1 year. Interestingly, these changes were associated with loss of the entire BND compartment. By extrapolation from parallel studies of in diabetic NOD mice, we believe that under these circumstances B cells relocalize in tissues rich in their autoantigen, where they become activated and can present antigen to T cells. These findings suggest that environmental events such as infection or injury may, by disrupting B cell anergy, dispose individuals to autoimmunity, the precise tissue target of which is specified by genetic risk factors.
-
Martha Campbell-Thompson, Dirk Homann & Richard Benninger
University of Florida
Funded by: Breakthrough T1D
Mapping the histopathological landscape of human T1D: a pilot study (CLARITY)The overall goal of this project is to extend studies examining human beta cells and immune cells in type 1 diabetes (T1D) to a new 3D fluorescent microscopy format for 500um or greater sized samples. These studies will adapt new tissue imaging methods (CLARITY) developed by Dr. K. Deisseroth (Nat Methods 2013). The specific aims are: 1. Adapt tissue transparency methods for pancreas sample preparation and multiple immunofluorescence staining. 2. Create robust imaging and data collection methods to produce 3D islet topographical maps. 3. Examine islets in situ from donors with prediabetes and established diabetes for evidence of β‐cell regeneration and death. 4. Characterize immune cell subtypes infiltrating islets and their expression of cytokines and chemokines.
-
Martha Campbell-Thompson, Lizette Duckworth-Vila, Ralph Hruban, Gunter Kloppel, Chen Liu, Nickolas Papadopoulos & Mark Atkinson
University of Florida
Diabetes and Pancreatic Cancers: Pilot study with molecular profilingOur study aims are the following:
1. Histopathology review of pancreata from patients without diabetes (no diabetes and autoantibody+) and
those within the diabetic and other groups (type 1, type 2, gestational, other (those with unclassified diabetes,
excludes cystic fibrosis)).
2. Molecular profiling of PanNET for neuroendocrine hormones and other genes (Isl1, p53, Ki67), FISH
(telomerase shortening), MEN1 mutations (DNA), and gene expression (RNA). Assays will be conducted at the
University of Florida and Johns Hopkins laboratories. -

David Carpenter, Miquel Porta & Sarah Howard
University at Albany
Persistent organic pollutants in the pancreatic tissue of people with and without diabetesThere is now strong evidence that persistent organic pollutants (POPs) such as polychlorinated biphenyls (PCBs) are major risk factors for type 2 diabetes. While the mechanisms involved are not known with any certainty, the associations between development of type 2 diabetes and blood concentrations of POPs are stronger than those for almost any other factor, including obesity. Some studies have suggested that obesity is in fact not a risk factor for type 2 diabetes, and that it is rather all of the POPs that are in the animal fats that are often consumed in excess that is really the risk factor.
However almost nothing is known about any possible involvement of PCBs and other persistent organics as factors contributing to type 1 diabetes. Type 1 diabetes is a quite different disease than type 2, but they do have some things in common. Our proposal is to begin investigation of the possible role of POPs in type 1 diabetes by obtaining measurement of concentrations of PCBs in the pancreas of individuals with type 1 diabetes as compared to controls and those with type 2 diabetes. PCBs will be used as an indicator POP. All POPs are fat soluble and are found almost exclusively in the fat component of human tissues. The pancreas, like every other organ, has some fat cells, and there are fats in the membranes of the beta cells that normally produce insulin. One possibility is that there is excessive accumulation of POPs including PCBs in the pancreas in causes of type 1 diabetes. This will be tested by direct determination of the PCB concentrations in tissues obtained through the nPOD project.
-

Gary Cline & Richard Carson
Yale University School of Medicine
Imaging pancreatic beta-cells with PET neuroimaging agentsA clinically viable means to measure pancreatic beta-cell mass (BCM) is essential for evaluating the physiological basis of therapeutic approaches to restore deficient insulin secretory capacity. Major advances in imaging BCM have been made by taking advantage of receptor-specific imaging probes that have been successfully used for neuroimaging. Hence, Positron Emission Tomography (PET) imaging ligands that were originally developed to specifically bind to neurons may prove useful for imaging BCM.
The Yale PET Center has been at the forefront of imaging both brain receptors and BCM and proposes to evaluate its extensive library of human neuroimaging agents as in vivo probes to quantitatively determine BCM. To maximize the value of these studies, pancreatic imaging in humans will be obtained together with validation studies in healthy and type 1 and 2 diabetes mellitus pancreas. Imaging probes that show suitable in vivo specific uptake in pancreas and appropriate imaging properties in humans, and can distinguish between healthy and diabetic pancreas in vitro will then be tested in a limited clinical PET-imaging trial to assess whether there is a measurable decrease in radiotracer binding in the pancreas of T1DM patients.
In Specific Aim 1, we will evaluate whether the approved radiotracers currently in use at the Yale PET center for human neuroimaging can be used for imaging BCM in healthy individuals.
In Specific Aim 2, we will validate β-cell specificity of the radioligands in vitro in healthy, T1DM, and T2DM human pancreas tissue obtained from the network for Pancreatic Organ Donors with Diabetes (nPOD), and in human islets.
In Specific Aim 3, we will perform a clinical evaluation in healthy and T1DM volunteers of those agents that meet the criteria of 1) promising pancreas imaging characteristics determined in Aim 1 and 2) favorable β-cell specificity as determined in Aim 2. A limited clinical evaluation is necessary to establish
-

Raphael Clynes
Columbia University
Funded by: NIH, Breakthrough T1D, and NIDDK
TCR signal transduction in diabetogenic T cellsTyrosine kinase inhibitors (TKis) effectively limit autoimmunity in animal models and are under clinical study in RA, MS and psoriasis. Recent studies in NOD mice have demonstrated proof-of-principal for promiscuous PTK blockade, using inhibitors initially developed to inhibit oncogenic pathways in cancer. Our recent work has shown that the selective Syk inhibitor R788 limits diabetes progression in NOD mice. Spleen tyrosine kinase (Syk) is most widely known as the PTK downstream of the B cell antigen receptor (BCR) and the IgG receptor (FcR) in myeloid cells and Syk PTKis have demonstrated clinical activity in RA and ITP. R788-treated NOD mice had decreased autoantibody levels presumably the result of BCR-inhibition and decreased T cell priming likely due to syk inhibition in DCs. A direct effect of Syk inhibtion on T cells was not reported, since syk expression in T cells was not expected. However, aberrant Syk expression may occur in autoimmune T cells. Indeed in our preliminary studies, discussed below, Syk is seen expressed in autoaggressive CD8 T cells infiltrating the skin and in activated murine and human T cells ex vivo.
Targeting of T cell activation via small molecule interference with protein tyrosine kinases is a promising approach to autoimmunity and could be directed at TCR signaling or co-stimulatory pathways. In T1D, these efforts will require determination of which signal transduction targets are specifically expressed in diabetogenic T cells in the islet and pancreatic lymph node. Autoaggressive T cells responding to sustained autoantigen drive may express signal transducing molecules not ordinarily expressed in resting or transiently active normal T cells thus providing attractive therapeutic targets. Our preliminary data shows that the Syk kinase is expressed in persistently activated T cells in vitro and in autoimmune T cells in vivo.
-

Ken Coppieters, Ditte Tornehave & Charles Pyke
Novo Nordisk A/S
IL-21/IL-21R and GLP-1R staining on pancreas tissues from (pre-)T1D, T2D and control donorsOur aim is to assess the protein expression level in conjunction with histological and (sub)cellular localization of the cytokine IL-21, its receptor IL-21R and GLP-1R in normal controls, (pre-)type 1 and type 2 diabetics (with history of incretin treatment for GLP-1R). (adopted by nPOD staff)
-

Barbara Coulson
The University of Melbourne at the Peter Doherty Institute for Infection and Immunity
Funded by: Agency: Australian Government-National Health and Medical Research Council
Analysis of type 1 interferon responses to rotavirus in patients with type 1 diabetesRotavirus infection of children at-risk of type 1 diabetes is associated with disease progression. Similarly, infection of adult non-obese diabetic (NOD) mice with Rhesus monkey rotavirus RRV accelerates diabetes onset. This acceleration correlates with RRV spread to the mesenteric and pancreatic lymph nodes, where virus associates with and activates dendritic cells and induces B and T cell activation. We also have shown that RRV exposure ex vivo induces generalized B and T cell activation by triggering toll-like receptor 7 (TLR7) signaling and type I interferon production by plasmacytoid dendritic cells (pDC) from NOD mice. As this activation includes islet autoantigen specific T cells, we hypothesize that type I interferon-mediated bystander activation contributes to diabetes acceleration by RRV in NOD mice. Notably, RRV induces significantly more activation of NOD mouse than C57BL/6 mouse cells, providing evidence that these responses to RRV are heightened in mice at-risk of T1D. Influenza virus and coxsackievirus B, which also signal through TLR7, have been shown to induce heightened pDC-dependent type I interferon responses in PBMCs from T1D patients compared to controls. We hypothesize that rotavirus will also induce heightened pDC-dependent lymphocyte activation and type I interferon secretion in PBMCs and other immune cells from patients with T1D compared to controls. Therefore, the overall aim of our study is to evaluate activation and type I interferon expression following stimulation with rotavirus in immune cells from patients with T1D compared to controls.
-

Nika Danial & Ulupi Jhala
Harvard Medical School
Funded by: NIH-HIRN
Molecular signatures of islet inflammation in type 1 diabetesThe process of β-cell decline in the lead-up to the overt diabetes could provide critical clues for timely therapeutic intervention. However the complexity, cell-type specificity, rapidly changing dynamics and the cumulative effects of inflammation all inform the outcome of the autoimmune attack on beta cell.
This proposal is designed to use multiple in vitro models systems of beta cell inflammation, along with human tissue samples as well as human serum samples from T1D children, to arrive at a temporal proteomic and metabolomics profile of human islet inflammation and death. These data are likely to contribute towards our understanding of biomarkers that would assist in staging the progression of human type 1 diabetes. The ultimately goal is to be able to use the information gleaned from the grant to identify windows of opportunity when therapeutic intervention could preserve beta cell mass and function to delay the progression towards full-blown diabetes.
-

Howard Davidson
University of Colorado Health Science Center
Funded by: Breakthrough T1D
Characterization of autoantigen-specific T cell receptors from PLNsType 1A Diabetes mellitus (T1D) is a predominantly T cell mediated autoimmune endocrine disease. Both CD4+ and CD8+ T cells reactive to diabetic autoantigens can be identified at low frequency in the peripheral blood of diabetic subjects and their at-risk relatives, but the extent to which these peripheral responses reflect those in the pancreatic draining lymph nodes (PLN) and insulitic lesions is currently unclear, mainly due to the relative inaccessibility of these tissues in living subjects. Answering this question is of central importance to the development of mechanistically relevant biomarker assays for staging disease and monitoring therapeutic efficacy, as well as for identifying appropriate targets for antigen specific immunotherapy. The goal of our project is to isolate and characterize T cells reactive to the major T1D autoantigens (with special focus on insulin, GAD65, IA-2, IGRP, and ZnT8) from the PLN and spleen of nPOD donors. The frequency, phenotype, epitope specificity and clonal diversity of islet-reactive T cells from these tissues will be compared with those present in islets and peripheral blood from the same donors (when available), and in PBMCs from HLA-matched living subjects at various stages of disease.
-

Mark Davis & Yu Wong
Stanford University
Funded by: Breakthrough T1D and Howard Hughes Medical Institute
Analysis of the antigen-specific T cell repertoire in T1D patientsThe majority of human T cell antigens relevant to T1D have been defined using algorithms which predict high affinity peptide binding to MHC. However, direct evidence showing that these antigens are presented to T cells by human islets and APCs in T1D patients has not been established for the vast majority of these peptides. A major goal of our project is to identify T cell peptide antigens relevant to T1D which meet the following criteria: 1) the peptides are naturally processed and presented by human islets/APCs and 2) there are T cells specific for these antigens in the PLN and/or islets of T1D patients. To achieve this goal, we are eluting peptides from class I MHC on HLA-A2+ human islets and identifying them by mass spectrometry to define naturally presented antigens. We are also verifying natural antigen presentation by using antigen-specific T cell lines as “biosensors” of antigen presentation on islets and APCs. Islet-specific peptides that are shown to be naturally presented will be used to make MHC-peptide tetramers and analyzed by CyTOF technology to measure the frequency, phenotype, and function of T cells specific for these antigens in T1D pancreatic draining lymph nodes obtained from the nPOD tissue bank. By verifying the natural antigen presentation of islet-derived peptides and the presence of activated T cells specific for these peptides in T1D patients, we will be able to differentiate between peptide antigens with bonafide relevance to T1D from islet peptides that are irrelevant to T1D. CyTOF analysis of the frequency, phenotype, and function of pathogenic T cells specific for disease-relevant islet antigens in T1D patients should be informative with respect this disease, potentially enabling sophisticated and early diagnoses and the ability to assess the efficacy of therapeutic intervention in human patients.
-

Marc Donath, Thierry Nordmann & Elise Dalmas
University Basel, Switzerland
Identification of islet associated immune cells in type 2 diabetic patients and further exploration of their role in disease progression and severityGrowing evidence suggests inflammation as a major underlying mechanism in the pathogenesis of type 2 diabetes mellitus (T2DM). Supporting the involvement of the immune system in T2DM, cross-sectional and prospective studies have associated elevated circulating levels of acute-phase proteins. Furthermore, clinical studies have demonstrated that anti-inflammatory drugs may improve glycaemia. Morphological and therapeutic intervention studies have uncovered an inflammatory process in islets of patients with type 2 diabetes characterized by the presence of cytokines, macrophages, β cell apoptosis, amyloid deposits, and fibrosis. However, the immune cells involved in pancreatic islet inflammation during T2D are insufficiently described. In human tissue samples, only CD68 positive macrophages have been documented. The level of activity of these cells as well as the occurrence and role of other immune cell types remain to be investigated.
Using the pancreatic tissue samples supplied by nPOD, we plan on investigating the presence of innate and adaptive immune cells within or around the islets of T2DM patients versus healthy donors. Defining the inflammatory milieu within the islets of T2DM patients will help us target further inflammatory treatments.
-

Dieter Egli, Bjarki Johannesson & Ruddy Leibel
Columbia University
Funded by: Helmsley Foundation
Generation of pluripotent stem cells from diabetics ADDENDUM: Generation of iPS cell lines from a majority of T1D casesORIGINAL PROJECT: Established different skin fibroblast cell lines, both from control and T1D nPOD donors. Graciously offered this as a resource available to current nPOD investigators.
ADDENDUM: The pancreata of nPOD cases are some of the best characterized type 1 diabetes cases. The availability of beta cells from stem cells would greatly enhance this resource by providing the ability to both study the primary pancreas, as well as living beta cells or other cell types in a defined environment with the genotype of the patient. This project aims to generate cell lines for as many of the nPOD cases as possible, and to convert selected cell lines to iPS cells, and stem cell derived beta cells to determine whether disease phenotypes are cell-autonomous effects of the genotype, or acquired through interactions with the environment or with other cell types.
-
Dieter Egli, Ruddy Leibel
Columbia University
Beta cell defects in cystic fibrosis related diabetesThe aim of this study is to determine how CFTR mutations affect beta cell performance, and whether
their effect is direct (cell-autonomous), or indirect, by affecting other cell types/physiological
processes with secondary consequences for beta cell function and viability. It is likely that any
mechanistic insights obtained from the study of this specific etiology of diabetes will be applicable to
other types as well. -

Garry Fathman
Stanford University
Microarray analysis of PLN from autoantibody positive donorsAs with many autoimmune diseases, T1D is initiated by some unknown inciting event (or events) that results in the appearance of disease-relevant biomarkers. Serum autoantibodies to islet antigens or changes in whole blood gene expression that appear before the onset of glucose intolerance can serve as biomarkers to identify pre-diabetic individuals. As we enter the era of intervention trials to block progression of T1D before beta cell destruction, it is important and beneficial to identify relevant biomarkers that can be used to stage early disease progression in T1D. Recent studies from my lab have demonstrated that changes in gene expression of pancreatic lymph node (PLN) tissue samples from AA+ individuals provided by nPOD, can be seen before the onset of abnormal glucose tolerance. These data also demonstrated that gene expression in the PLN is similar between T1D patients who have had hyperglycemia for two or more years and normal subjects, suggesting that the inflammation accompanying progression to hyperglycemia in AA+ subjects can be identified by changes in gene expression in AA+ subjects, and may be used to identify biomarkers, and/or suggest targets of, or response to therapy. Because PLN tissues cannot be readily biopsied from AA+ individuals, our rationale is to examine gene expression in PBC samples provided by the TrialNet Natural History tissue repository to ask if there are distinct changes in gene expression seen in the PBCs (similar to those seen in the PLN) that occur with the progression of disease. As a second project arising from these data, we will ask if there are changes in gene expression that accompany intervention therapy with CTLA4-Ig in a TrialNet intervention trial being run by Dr. Bill Russell at Vanderbilt. These approaches may help identify early biomarkers of disease that will allow sub-setting of AA+ patients based on stage of disease or rate of progression, and could identify prediction of, or response to, therapy.
-

Brian Fife & Marc Jenkins
University of Minnesota
Funded by: Minnesota Partnership for Biotechnology and Medical Genomics, Breakthrough T1D and NIH
Tracking islet autoantigen specific CD4+ T CellsThe goal of this research project is to identify self-reactive T cell clones and monitor the frequency and activation state during the progression of autoimmune diabetes. Identification of T-cell reactivity to beta-cell antigen epitopes is important to study pathogenesis and monitor antigen specific interventions in T1D. We have successfully generated and validated human tetramer reagents to diabetes relevant target antigens to determine frequency and activation state of autoreactive CD4+ T cells in patients with diabetes. Tetramer reagents could provide much needed biomarker reagents to predict T1D earlier in ‘at risk’ patients, stage clinical disease, and determine if therapeutic approaches have an effect on the CD4+ T cell level during T1D pathogenesis. If successful, the tetramer biomarkers and enrichment approach we are testing could precede autoantibody production and provide an earlier diagnosis of T1D, before irreparable damage has occurred to the islets. Therefore, these reagents would have great impact to provide a new in vivo diagnostic approach to identify T1D T cell responses as an early diagnosis for high risk patients. With a better understanding of beta cell specific targets we will be able to develop novel therapeutic approaches to cure diabetes.
-

Paolo Fiorina & Francesca D'Addio
Boston Children's Hospital
Role of TMEM219 Expression in Type 1 DiabetesThe recent failure of immunotherapeutic strategies in the cure of T1D raised questions in considering autoimmunity the sole mechanism responsible for the pathogenesis of T1D. It has been recognized that activation of immune response against self-peptides is essential in the onset of T1D, but its role in the destruction of beta cells, which leads to reduced insulin secretion and glycometabolic control, is a matter of debate. The recent studies demonstrating a relevant role for the environmental factors in the pathogenesis of diabetes, confirmed that non-immunological determinants and beta cell regulating molecules might contribute in initiating and/or perpetrating this process. The increasing body of evidences that autocrine and paracrine signaling pathways control the homeostasis of several type of cells, such as stem cells and pancreatic beta cells, drew the attention of scientists on the possibility that even circulating hormones may be involved. More importantly, altered secretion of factors/hormones during T1D leads us to hypothesize that systemic factors may target pancreatic beta cells and modulate fate/function. Our previous studies showed that a circulating hepatic-released factor IGFBP3, exerts a detrimental effect on intestinal stem cells (ISCs) and induces their apoptosis/death by binding its receptor, TMEM219, expressed on ISCs surface. Interestingly, TMEM219, is expressed in human islets and in a beta cell line. We hypothesize that an overexpression of TMEM219 may favor beta cells destruction and affect beta cell mass, and the consequent hyperglycemia may perpetrate the process, given that TMEM219 expression is sensitive to high glucose exposure.
-

Gun Frisk & Olle Korsgren
Uppsala University, Sweden
Funded by: EU and Diabetesförbundet
Different expression of extracellular CAR in islets in pancreatic sections and isolated isletsThe etiology of the type 1 diabetes (T1D) is unclear but it has been shown that genetic factors influence the pathogenesis. In addition, several studies have shown that environmental factors likely contribute to the disease development. One such factor is virus infections, particularly the Coxsackie B viruses (CBVs), are among the main candidates and there has been numerous of studies showing association between these gut viruses and T1D. To be able to infect a certain cell it has to express an appropriate receptor and the main receptor for CVBs is the tight junction protein Coxsackie-Adenovirus-Receptor (CAR). The expression of the CAR protein has been shown to be increased during healing and during inflammation. The primary aim of this study is to determine the expression of CAR in pancreas especially on b-cell, and to what extent this expression is affected by the disease progress in T1D. The expression on human isolated islets, infected and un-infected cultured for prolonged periods will be studied also be studied. CAR expression in pancreas will also be studied in human pancreatic tissue from recent onset T1D donors, long standing T1D donors, islet autoantibody positive donors, T2D donors and controls to see if this protein “normally” is expressed in pancreas, especially on the b-cells. Studies are also on-going to see if the expression is affected by the auto- immunity in the pre diabetic and T1D islets. Preliminary results indicate that the expression level of CAR is significantly higher in islets from diabetes related donors compared to controls. It is also 10-100 fold higher expression in the endocrine tissue from pre diabetic and T1D donors compared to the exocrine tissue from the same donors.
-

Michael German & Chester Chamberlain
University of California San Francisco
Funded by: The Caring for Carcinoid Foundation
Analysis of normal pancreas from PNET patients with and without diabetesSpontaneous recovery from established type 1 diabetes (T1D) is so rare that only one well-documented case has ever been described in an adult, and in that case recovery resulted from an insulin-secreting tumor, not regeneration of the patient’s normal beta-cells. Therefore, when we saw a 50 year old man at UCSF with T1D who got off insulin (Patient 36300562), we investigated the cause. He had a pancreatic neuroendocrine tumor (PNET), but it was not producing insulin. Instead, in the few islets in his pancreas that still contained beta-cells, those beta-cells were proliferating at rates far exceeding anything previously reported in humans, suggesting that PNETs may produce factors that can promote human beta-cell regeneration. We also found alpha-cells of Patient 36300562 were producing ghrelin, suggesting that gut hormone expression in islet cells may be a characteristic of the T1D pancreas. SPECIFIC AIM 1: Characterize the expression of ghrelin in the pancreatic islets of patients with and without diabetes. We will perform immunostaining assays for ghrelin in sections of normal and T1D pancreas samples from the nPOD. We will also examine the expression of PYY and somatostatin to gain a better understanding of how hormone gene expression may be different in the islet of T1D patients. These studies will help clarify whether the expression of ghrelin in the alpha-cells of Patient 363000562, who had T1D, is a consequence of the PNET or T1D. These studies will also add to our knowledge of the T1D pancreas phenotype. SPECIFIC AIM 2: Characterize the beta-cell phenotypes in patients with PNET. The high beta-cell proliferation rate and T1D recovery of Patient 36300562 suggests that PNETs may produce factors that can promote human beta-cell regeneration. We will therefore also examine markers of beta-cell proliferation (Ki67) and death (TUNEL). These studies will help confirm the beta cell phenotype of Patient 36300562, and may help justify additional studies to identify novel factors from PNETs that promote human beta-cell regeneration.
-

Roberto Gianani
University of Colorado, Denver
Characterization of type 1 diabetes subsetsType 1 diabetes is a clinically and pathologically heterogeneous disease. A large subset of individuals with Type 1 diabetes has an autoimmune form of the disease characterized by subtotal loss of beta cells and positivity for islet autoantibodies as well an increase prevalence of DR3 and DR4. We have recently characterized a pathological pattern of beta cell loss in Type 1 diabetes that does not appear to be associated with autoimmunity. We hypothesized that this represent a novel disease characterized by “degenerative” loss of beta cells possibly due to increased apoptosis (affecting also alpha and delta cells), pathogenetically distinct from the beta cell loss seen in Type 2 diabetes.
The Specific Aims Are:
- Morphometric characterization of Pattern B type 1 diabetes in relation to the beta cell, alpha cell and delta cell relative volume as compared to controls and type 2 subjects.
- Investigation of surviving expression at the protein and RNA level (initially these studies will be limited to surviving).
- Comparison of apoptosis rate between different groups
- Investigation of amyloidosis in the different groups
- Collaborative efforts with Dr. Sechi Laboratory.
-

Nick Giannoukakis & Mark Atkinson
University of Pittsburgh
Role of bacterial biofilms in the onset of pancreas inflammation in T1DEven though the immunopathogenic triggering event(s) in type 1 diabetes (T1D) remain enigmatic, there is accumulating evidence, mainly in rodent models of the disorder, suggesting that pancreas-resident antigen-presenting cells (APC) are activated very early post-natally. This results in the recruitment of innate immune cells like neutrophils into the pancreas even before adaptive immune cells that are islet antigen-specific are detected. Activated neutrophil accumulation into new-onset T1D human pancreata has also been documented. While it is possible that this innate immune activation very early-on reflects a reparative response to some endogenous natural post-natal “remodeling” process gone-astray, or to the incursion of viruses, the direct associations have not been conclusively established. The kinetics of the activation of resident APC and the post-natal aggravation of innate immunity prior to any signs of T-cell incursion is better associated with pattern recognition receptor-mediated signals, implying a bacterial response. Indeed, the NOD/ShiLtJ mouse strain as well as human T1D patients, have been shown to exhibit a “leaky gut” phenomenon and treatment of NOD mice with antibiotics or an exchange of the gut microbiota has resulted in prevention of disease onset. Furthermore, microbiota from the “leaky gut” have been shown to localize inside the pancreas, triggering an inflammation. In spite of recent studies aimed at identifying specific microbes as triggers, there is no evidence pointing to a specific pathogen. Our hypothesis, instead, proposes that any bacterial pathogen can trigger the resident APC through the formation of biofilms. Bacterial biofilms are omnipresent in tissues in which bacteria reside and act not-too-unlike extracellular trap structures. These highly pro-inflammatory microenvironments compel tissue repair and remodeling. Biofilm constituent molecules not only protect and promote the survival of entrapped bacteria, but also act as “danger” signals to the pancreas-resident APC. Biofilm formation, consequent to post-natal “leaky gut”-facilitated bacterial deposition inside the pancreas is hypothesized to trigger the resident APC, to attract neutrophils and migratory macrophages and dendritic cells, provoking an inflammation that ultimately culminates in the activation of autoreactive lymphocytes inside the pancreas-draining lymph nodes. The objective of this study, using nPOD human pancreas tissue, is to first demonstrate the existence of bacterial biofilms inside the pancreas of T1D subjects. Then, to compare the incidence of biofilms in the T1D pancreata to non-diabetic pancreata and to determine any associations of biofilm presence, density, and “signature” with age-at-onset of disease. In parallel, we will determine if the presence of biofilms and/or specific signatures, is associated with the presence of particular innate immune cell populations and their state of activation in situ. To realise these objectives experimentally, we will deploy multiparameter immunofluorescence microscopy to identify specific immune cell populations; fluorescence in situ hybridization to identify biofilms, and sequencing technology to identify biofilm signatures in the pancreas tissues. On its own, we believe that this project is novel since it is the first-in-kind to explore the potential involvement of biofilms in organ-specific autoimmunity, and it can also inform a better understanding of the initiating trigger of T1D altogether.
-

Kathleen Gillespie
University of Bristol
Funded by: Diabetes UK
Maternal microchimerism in T1D pancreasMaternal microchimerism (MMc) is acquired by an infant during pregnancy and these cells are maintained in some individuals for decades. Encountering maternal antigens during pregnancy represents the first immunological challenge for the fetus although maternal cells are protected from detection by induction of suppressive fetal regulatory T cells.
In type 1 diabetes increased levels of maternal microchimerism have been demonstrated in peripheral blood DNA and maternal cells have been identified in human pancreas. The aim of this study is to phenotype these maternal cells in well characterised male pancreases in order to help understand how the presence of maternal cells could contribute to risk of T1D. The methodology used will utilise using X/Y chromosome FISH with concomitant immunofluorescence. Once the phenotype is established, we will attempt to confirm the maternal origin using single cell laser capture with DNA profiling.
-
Kathleen Gillespie & Abby Willcox
University of Bristol
Funded by: Diabetes UK
Molecular insights into the type 1 diabetes pancreasPancreatic lymph nodes (PLN) are the central location for the autoimmune response that leads to type 1 diabetes (T1D), yet no histological studies of this crucial step in pathogenesis of human diabetes exist. In this study, we request sections from T1D and matched control PLN to allow us to follow up our preliminary observations showing that replicating germinal centers are diminished in recent onset T1D. In particular, we will focus characterizing the structure and cellular content of PLN in T1D and will test the hypothesis that lack of germinal center replication reflects attempted regulation of the immune response.
-

Maria Grant
Indiana University
Bone marrow progenitor cell (BMPCs) dysfunction in diabetes is mediated by reduced bioavailability of NODespite advances in understanding the pathogenesis of diabetic retinopathy (DR), the nature and time course of the molecular changes associated with DR remain incompletely understood. We propose to investigate the mechanism of bone marrow (BM) failure in diabetes and test diverse therapeutic approaches to correct BM and bone marrow progenitor cell (BMPC) dysfunction in order to restore retinal vascular health. Healthy BMPC participate in the process of re-endothelialization and also contribute indirectly to vascular repair by the secretion of paracrine factors. In health, distinct populations of BMPC work in concert to orchestrate vascular repair. The small pool of CD34+ cells has robust reparative function and modulates, via paracrine signaling, the function of the much larger pool of CD14+ cells which, depending on the local milieu, have the potential to also become efficient reparative cells. Freshly isolated BMPC can be expanded in vitro to generate two additional populations of reparative cells: early endothelial precursor cells (eEPC) or late outgrowth endothelial cells (OEC) that may have more therapeutic utility than freshly isolated cells. To date the optimal progenitor cell for vascular repair has not been identified.
In diabetes, BMPC demonstrate deficient reparative function and may even have a deleterious effect on the repair process. We demonstrated that: 1) diabetic CD34+ cells do not repair acellular capillaries in the diabetic retina, whereas CD34+ cells from nondiabetic individuals do (a similar observation was also made using CD14+ cells); 2) compared to nondiabetic CD34+ cells, diabetic CD34+cells have reduced bioavailable nitric oxide (NO) which results in their reduced migratory ability that can be corrected by restoring NO levels to normal; and 3) diabetes is associated with the loss of circadian rhythmicity of BMPC release from the BM into the circulation. Decrease in blood flow through the vasa nervorum, the small arteries that provide blood supply to peripheral nerves, has been implicated in the development of diabetic neuropathy. We postulate that in diabetes reduced NO initiates changes in the vasa nervorum and is a mechanistic contributor to reduced sympathetic neurotransmission which ultimately results in BM dysfunction. Loss of this neurotrophic support leads to changes in the BM microenvironment which contribute to, not only reduced release of these cells, but also to their impaired function. This dysfunction is further exacerbated by elevated reactive oxygen species (ROS) in these cells. Based on our findings that diabetic retinopathy is linked to peripheral neuropathy, we put forth the following hypothesis:
Loss of circadian rhythmicity of release of BMPC from the BM (due to dysregulated NO in the vasa nervorum and subsequent BM neuropathology) and increasing levels of intracellular ROS, increasing endogenous NOS inhibitors and inappropriate secretion of pro-inflammatory cytokines and proangiogenic growth factors compromise the reparative ability of BMPC leading to development of diabetic retinopathy.
-

Dale Greiner & Leonard Shultz
University of Massachusetts
Diabetogenic function of autoimmune donor splenocytes in humanized miceOur understanding of type 1 diabetes (T1D) has been advanced greatly by studies carried out using mice and rats. However, rodents are not humans and progress in the understanding of the pathogenesis of T1D in humans has been impeded by the a lack of assays that permit the direct in vivo analysis of diabetogenic human T cell populations without putting individuals at risk. The availability of spleen, bone marrow, and thymus samples from T1D and islet autoantibody-positive donors provides an opportunity to analyze the diabetogenic function of these cell populations following engraftment in newly developed humanized mouse model systems. This work is based on our recently developed immunodeficient mice that express human HLA molecules and are optimized for human immune cell engraftment. Our goal is to use unique tissues available from nPOD to recapitulate autoimmune T1D in humanized mice. We will engraft human hematopoietic stem cells and immune cells into HLA-matched NSG mice and determine the ability of cells from T1D donors to attack islets and induce the destruction of insulin-producing cells. The knowledge obtained from these studies will permit us to elucidate the pathogenesis of T1D and to develop new approaches for preventing or curing the disease.
-
Dale Greiner & Rita Bortell
University of Massachusetts Medical School
Investigate the molecular link between increased risk of diabetes among patients with psychiatric disordersCompared to the general population, studies show 2-3 times increased risk of developing metabolic syndrome in drug naïve patients with severe mental illnesses like schizophrenia or bipolar disorder. Impaired glucose metabolism in non-obese never-treated patients and in first degree relatives of schizophrenic patients, suggests a possible genetic association between diabetes and schizophrenia. We are proposing to study the molecular cause of increased metabolic risk among patients with psychological disorders.
-

Zhiguang Guo, Alex Rabinovitch & Romano Regazzi
Sanford Research/Sanford Health
Funded by: NIH
Identifying and Validating Noncoding RNAs as Human Beta Cell-Specific BiomarkersDirect identification and validation of human β cell-specific biomakers is critical for monitoring the change of human β cell mass at prediabetes and the new-onset diabetes stage and during the therapeutic intervention. Due to technical and ethical constraints, our ability to prospective studies of human β cell biomarkers in response to β cell stress/apoptosis and its correlation with early β cell loss in human is limited. The objective of our study is to develop biomarkers of human β cell stress/apoptosis correlating with β cell loss that can be used to non-invasively monitor the development of diabetes pathogenesis and the outcome of treatments aimed at preserving or restoring functional β cell mass. The goal of this study is to identify potential biomarkers including a panel of noncoding RNAs in human islets that are correlated with β cell stress and apoptosis and to determine whether novel β cell biomarkers in circulation are correlated with early human β cell loss. We hypothesize that β cell ER stress and apoptosis induction increases a panel of noncoding RNAs in human islets in the pancreas of patients with diabetes and dead or dying human β cells can release them into circulation and the levels of these noncoding RNA in circulation are associated with β cell loss. We plan to collect islet cells for miRNA expression profiles by laser capture microdissection (LCM) and determine whether a panel of β-cell specific noncoding RNAs in islets of human pancreas specimens from patients with type 1 and type 2 diabetes are up-regulated by real-time PCR array. We will confirm some up-regulated noncoding RNAs in human islets are β-cell specific by miRNA fluorescence based in situ hybridization (FISH). We will validate this panel of noncoding RNAs, as β cell-specific biomarkers, in plasma of patients with type 1 and type 2 diabetes. Success in our proposed experiments would help us to further develop a panel of β cell biomarkers, which are correlated with early β cell loss, for clinical evaluation of disease stages including pre-diabetes and of therapeutic interventions to promote β cell health and survival.
-

Abdel Hamad & Thomas Donner
Johns Hopkins University
Fas Ligand: Unorthodox target for prevention of type 1 diabetesDespite improvement in insulin delivery, maintaining tight control of glucose homeostasis continues to be a challenge that results in bouts of severe hypoglycemia and hyperglycemia and serious long term complications in many patients. Therefore, developing an immunotherapy for type 1 diabetes (T1D) remains a major goal. Reaching this goal requires detailed knowledge of the diabetogenic process. Particular knowledge of the molecules that play pivotal roles in driving the pathogenic mechanisms but are not required for mounting normal immune responses could lead to uncovering of safe therapeutic targets. Fas ligand (FasL) is a rare example of such molecules. It is an apoptosis-inducing member of the TNF family that is not required for immune responses. Yet it plays a critical role in driving the diabetogenic process as non-obese-diabetic (NOD) mice bearing spontaneous loss-of-function mutation of FasL (gld) of Fas (lpr) are completely protected from the disease. We hypothesize that aberrant FasL-mediated apoptosis of IL-10-producing regulatory B cells is critical for initiating the diabetogenic process. This hypothesis is supported by strong published and unpublished data in the NOD mouse model. By working with Breakthrough T1D/nPOD, we are investigating whether a dysregulated Fas pathway is compromising the role of IL-10-producing regulatory B cells in humans. We have collected strong evidence, so far, indicating that, as in NOD mice, aberrant FasL-mediated apoptosis may a pathogenic mechanism in T1D. Such results will not only provide new insights into the disease process, but also a platform for novel therapeutic modalities that do not impair the patient host defense.
-

Manami Hara
University of Chicago
Funded by: Breakthrough T1D
Beta-cell destruction and preservation in early and late onset type 1 diabetesType 1 diabetes (T1D) is one of the most common chronic diseases of childhood with onset peaking between 5-7 years of age and around puberty. Early childhood-onset T1D often exhibits severe beta-cell depletion compared to late adult onset T1D. We propose to test the hypothesis that immature beta-cells in early childhood are more susceptible to autoimmune destruction, whereas the cell death of mature beta-cells in adult onset T1D undergoes different kinetics and its process is substantially slower than previously considered. A better understanding of distinct beta-cell destruction and preservation during childhood could lead to new therapeutic strategies to treat young children with T1D.
-

Paul Harris, Masanori Ichise & Matthew Freeby
Columbia University
Funded by: NIDDK and Breakthrough T1D
Determination of specific and non-specific binding of 18F-FP-DTBZ in whole pancreas homogenates obtained from controls and patients with longstanding type 1 diabetesIn the past we identified Vesicular Monoamine Transporter Type 2 (VMAT2), as a biomarker of beta cell mass that is quantifiable in vivo by Positron Emission Tomography (PET). PET is a tomographic imaging technique providing accurate non-invasive dynamic measurements of regional PET tracer uptake and clearance. These measurements are used to calculate VMAT2 density in the pancreas as a surrogate measure of beta cell mass. Immunohistochemical staining of both control and long term T1D pancreata show excellent correlation between VMAT2 and insulin staining. When PET and the radiopharmaceutical 18F-FP-DTBZ are used to non-invasively measure VMAT2 in vivo, we observe that healthy subjects have a significantly greater pancreatic VMAT2 signal than long term T1D patients with documented inability to secret normal amounts of insulin. However there is a significant residual VMAT2 signal in the pancreata of the T1D patients.
The goals of our project are to understand the nature of this background signal. There are three not mutually exclusive explanations of the background signal observed in pancreata believe to be beta cell ablated due T1D; 1) The radioligand 18F-FP-DTBZ is binding specifically to VMAT2 or some other receptors, not present in beta cells (specific and displaceable off-target binding). Specific off target binding is being measured in a clinical PET study currently in progress. We have also quantified, using immunohistochemistry, the expression of VMAT2 laying outside the beta cell subset in human pancreata (e.g. PP cells). The specific off target bind does not seem to account for the background, 2) 18F-FP-DTBZ may also bind non-specifically and non-displaceably to pancreas tissue (off target non-specific binding). The in vivo non-specific binding is currently being measured in clinical PET study in progress. Our in vitro estimates of non-specific binding using cadaveric pancreas tissue supplied by NPOD suggest that a significant portion of the in vivo background signal is due to non-specific binding. However, these in vitro methods may overestimate in vivo binding of the tracer and must be interpreted with caution, 3) The last formal possibility, that our measurements of VMAT2 in the long term T1D pancreas indeed are on target and specific, representing residual VMAT2 possibly associated with beta linage cells is now under consideration. Such a possibility seems to conflict with preexisting immunohistochemistry data, but immunohistochemistry is not the most sensitive technique available for detecting low levels of expressed proteins. The next phase of our project will be to study long term diabetes pancreata for VMAT2 gene expression using in situ hybridization and RT-PCR techniques. While the pancreas is a difficult organ to obtain RNA suitable for such studies, the generally excellent quality of NPOD tissues will improve our chances for success.
-

Matthias Hebrok & Long Cai
University of California, San Francisco
Funded by: Breakthrough T1D
Investigating the de-differentiated state of the β-cell in human diabetic patient tissuesIt is well accepted that compromised ß-cell function is an integral part of diabetes development. Several recent studies, mostly using complex genetic mouse models, have supported the notion that a change in the ß-cell state or identity due to varying insults causes diabetes in the absence of ß-cell death. Our work has focused on the involvement of the von Hippel Lindau/Hypoxia Inducible Factor (VHL/HIF) pathway, a master regulator of cellular response to diminished oxygen (hypoxia). In a mouse model that stabilizes the HIF pathway despite normal oxygen levels within the ß-cell, we observe the development of diabetes which correlates strongly with a) a loss of the ß-cell differentiation state as determined by markers such as Pdx-1, Nkx6.1, MafA, Glut-2, and Insulin, and b) inappropriate activation of embryonic signaling pathways and genes. Immunofluorescence analyses on islets from diabetic animals displayed strikingly reduced level of insulin as compared to control, normoglycemic littermates, which indicate the cause of diabetes to be insulin insufficiency. The broad disruption of gene expression observed in diabetic mice suggests a loss of ß-cell identity (or dedifferentiation) as the underlying cause. We have also shown that ectopic expression of a key transcription factor Sox9 (normally expressed during embryogenesis) in the ß-cell causes dedifferentiation and diabetes. Our goal is to translate our findings from the mouse models of diabetes into the human condition. It is becoming apparent that diabetic patients can be stratified into distinct cohorts (based on onset of disease, age, weight, race) and it is critical to identify what sub-population of patients develops the disease due to ß-cell dedifferentiation. To this end, samples available from the nPOD provide an invaluable resource to a) identify patients who have compromised ß-cell identity, and b) correlate the findings from mouse models into human patients by identifying what signaling pathways or genes are erroneously regulated in these samples. Mouse tissue allows us to conduct genome wide expression analyses that can lead to identification of important regulators to be tested in the human samples. Our focus is on the maintenance of identity of the ß-cell, and discovering important regulators of this process may enable the development of intervention therapies that could block or delay the onset of ß-cell damage.
-
Kevan Herold & Alfred Bothwell
Yale University
Funded by: Breakthrough T1D
Reconstitution of HHLS mice with bone marrow from patients with T1DMThe focus of our Breakthrough T1D Autoimmunity Center is to create and study a humanized mouse model of Type 1 diabetes. The approach that we are developing involves use of immune deficient (γc-/-) mice with “knock-in” of human cytokine and other genes that are needed for optimal reconstitution with human cells. In addition, we have created transgenic mice that express human proinsulin in beta cells and in addition, have human MHC+ mouse MHC- mice in which the presentation and responses to human antigens (e.g. proinsulin, insulin) can be studied. We hope to reconstitute the immune deficient mice with human CD34+ bone marrow cells from patients with diabetes. The duration of diabetes should not be an issue so the use of nPOD tissue would be appropriate. We plan to study the ability of the reconstituted mice to develop diabetes – spontaneously or with provocateurs such as low dose streptozotocin, immunization with antigens, or even transgenic expression of B7.1 on beta cells. (adopted by nPOD staff)
-

Akihisa Imagawa & Toshiaki Hanafusa
Osaka University, Japan
Funded by: Gift Fund
Histological differences between Japanese and Western type 1 diabetes(1) To clarify the infiltration of mononuclear cells in exocrine pancreas of Western type 1A diabetes and compare the histological findings to those of Japanese patients
(2) To clarify the expressions of the molecules related to innate immunity or viral infection in Western type 1A diabetes and compare the hitological findings to those of Japanese patients
-

Dolores Jaraquemada, Mercè Martì & Carme Roura-Mir
University of Barcelona, Spain
Tracing effector and regulatory T cell populations in type 1 diabetesOur group is studying T cell phenotype and antigen presentation and recognition by T cells in T1D, with three main objectives. (i) The characterization of role of T cells involved in immunoregulation, i.e. regulatory T cells (nTregs or iTregs) and NKT cells. We are studying the mechanisms of regulation by analyzing the role of soluble and membrane bound molecules on the regulatory effect of each cell type; and the cooperation between regulatory populations to achieve a higher degree of suppression of T effector cells. (ii) The characterization of the TCR repertoire both on the pancreas infiltrate and on PBMCs from T1D patients. TCR expressed by autoreactive T cells is important as a tracer of the different clonotypes involved in T1D pathogenesis. We are also working on the hypothesis that some of the T cells involved in the maintenance of the chronicity share the TRBV-CDR3 as public TCRs. If proven, TCR repertoires prone to autoimmunity could be defined when accompanied with the HLA-associated alleles. Expanded iNKT cells will be analyzed as a model of public TCR in those patients. (iii) The third aim addresses antigen recognition by autoreactive T cells by the characterization of peptides presented in situ by HLA molecules. We are now able to retrieve a good number of peptides from small samples (i.e. islets). We will compare the T-cell epitopes already identified in T1D with those naturally presented in the islets by HLA class I and class II molecules. Because of the low number of HLA-II positive cells in the islets, we expect results for class I and are working on alternative approches for class II.
The first part of this project was in part funded by the Breakthrough T1D and we are now funded by the Spanish Department of Science.
-

Jonathan Katz
Cincinnati Children’s Research Foundation
Manipulating DNA damage-response signaling for the treatment of type 1 diabetesAs T cell toggle through their various life cycle, certain distinct biological changes occur that can be specifically targeted with small molecule drugs to target these cells for cell death. This is particularly true of activated autoreactive T cells. We have developed a combination of small molecular drugs that can target activated T cells and push these cells into apoptosis during (re-)activation. We propose here to test if these drugs can specifically target islet reactive T cells from T1D patients and pre-diabetic individuals while specifically sparing Treg and T memory cell subsets.
-

Seung Kim
Stanford University
Funded by: HHMI
Investigating islet alpha-to-beta cell conversion in DiabetesDiabetes occurs when insulin producing pancreatic β-cells are impacted: type 1 diabetes due to destruction of β-cells, and type 2 diabetes due to a decrease in their number and/or potency. Consequently, therapy that aims to replace β-cells is a promising long term alternative to the currently prevalent insulin injections. However, the difficulty in finding viable sources to generate functional β-cells remains a crucial bottleneck. Herrera and colleagues recently discovered that under conditions of near-total depletion of β-cells in mice, only α-cells within pancreatic islets spontaneously convert into β-cells1. Encouragingly, there is in fact an overabundance of α-cells in the pancreas of diabetes patients. However, today we lack the knowledge to initiate and maintain α-to-β-cell conversion and are thus unable to exploit the availability of abundant α-cells to orchestrate this process reproducibly in humans.
Exciting preliminary results from our laboratory and others reveal that a subset of pancreatic α-cells in T1D juvenile donors lose α-cell gene expression and acquire the expression of several key β-cell genes including Insulin. These data suggest that α-to-β-cell conversion may have occurred in these T1D donors. Further, we identified genes that likely control α-cell fate by recapitulating our T1D findings in mice that were engineered to delete specific genes (Dnmt1 and Arx) in their α-cells. The work proposed here aims to extend our findings in multiple T1D donor samples with long or short disease durations. To our knowledge, the question of whether α-to-β-cell conversion occurs in T2D has not been previously studied.
We therefore request pancreas samples from T2D donors to analyze them for evidence towards α-to-β-cell conversion. We will use state-of-the-art technologies to enable us to isolate α-cells undergoing conversion and probe them for changes in their genetic and epigenetic landscape. These studies are expected to a) Elucidate the similarities and differences between the fates of pancreatic α-cells in early and late stage T1D, and between T1D and T2D, and b) Identify new molecules and pathways that can be targeted for developing drugs to enhance the generation of functional β-cells.
-

George King & Susan Bonner-Weir
Harvard University
Funded by: Breakthrough T1D
Joslin Medalist StudyHistory of the Joslin Medalist Program
Joslin first began awarding medals to people with diabetes in 1948 with a 25-year Victory Medal. Believing that proper self-management was the key to minimizing long-term complications, the program was the vision of Elliott P. Joslin, M.D. and served as an incentive for those committed to good, though challenging, diabetes care. In the early 1950s the name was changed to the Blue Ribbon, and as more and more people lived long healthy lives with diabetes it finally became the 25-year Certificate that is awarded today. In 1970, Joslin expanded the program and began awarding a 50-year bronze medal. And Joslin presented the first 75-year medal in 1996.
50-Year Medalist STudy
Joslin’s 50-year medalists are invited to participate in a special study, examining outcomes of long-term diabetes. The study attempts to understand what factors contribute to the longevity of individuals who have received this honor. Currently, over 400 medalists are interested in participating in the study. To date, over 300 of these have completed an extensive questionnaire about their life with diabetes. Data from this questionnaire suggest that the risk of kidney, eye, and nerve problems is different after 50 years with type 1 diabetes than the risk among all individuals with the disease.
A second study is currently being undertaken, which examines factors in the blood and DNA that may help in modifying the risk for complications and survival. Participation in this study is open to all individuals residing in the United States who have received the Joslin 50-Year Medal. In coming months, updates for this study and findings from the previous study will be posted on a separate Web site.
-

Richard Kitsis
Albert Einstein College of Medicine
ARC, a novel beta cell survival factor in type 2 diabetesThe progression of type 2 diabetes is mediated to a large extent by the deaths of β-cells, the cells in the pancreas that synthesize and secrete insulin. We discovered that ARC (Apoptosis Repressor with CARD), an endogenous inhibitor of cell death, is expressed at high levels in β-cells of the mouse pancreas. Using cell culture studies and genetic mouse models, we have further demonstrated that ARC functions as a critical β-cell survival factor during type 2 diabetes. However, little is known about the expression patterns of ARC in human β-cells and other cells within the endocrine pancreas. This information is critical for understanding the role of ARC in the pathogenesis of diabetes in humans. We have assessed whether ARC levels in islets decrease during diabetes, and found no significant changes in ARC abundance in old versus young ob/ob mice. Unexpectedly, however, we observed that the subcellular localization of ARC differs in diabetic versus non-diabetic mice. In this study, we propose to characterize ARC expression in islet cells of human pancreatic tissue from non-diabetic individuals and those with type 2 diabetes.
-

Mitchell Knutson
University of Florida
Funded by: NIDDK and NIH
Molecular mechanisms of iron uptake by pancreatic beta cells and their contribution to the development of diabetesHuman disease states that result in excess iron accumulation, e.g., hereditary hemochromatosis and beta thalassemia major, are associated with an increased incidence of diabetes. In these disorders surplus iron overwhelms normal transport and storage mechanisms leading to the appearance of excess free iron in circulation and the subsequent deposition of iron in tissues such as the pancreas. Excess iron in tissues can lead to the formation of free radicals, which can damage beta cells and lead to cell death or dysfunction. Despite the long-established connection between iron overload and diabetes, exactly how iron is transported into pancreatic beta cells is not yet known. Currently only three mammalian proteins are known to be able to transport free iron: Divalent Metal-Ion Transporter-1 (DMT1), ZRT/IRT-Like Protein 14 (ZIP14), and ZRT/IRT-Like Protein 8 (ZIP8). The aim of our project is to determine the expression and subcellular localization of these established iron transporters in the cell types of the human pancreas, focusing primarily on beta cells. Identification of potential routes for beta cell iron uptake will provide targets for future mechanistic studies concerning the relationship between beta cell iron loading and function.
-

Jake Kushner
McNair Interests
Do de-granulated ß-cells persist in type 1 diabetes?The goal of this proposal is to define the lineage mechanism of ß-cell persistence in type 1 diabetes (T1DM). A few ß-cells often persist in T1DM pancreata. But, the lineage mechanism of ß-cell persistence in T1DM remains poorly understood. Our overarching hypothesis is that ß-cell persistence in T1DM is due to ongoing ß-cell regeneration. Alternatively, ß-cell persistence in T1DM could be due regional differences in islet autoimmunity, or because ß-cells acquire a degranulated state that protects them from autoimmune mediated destruction. These hypotheses are readily testable with NPOD samples. However, by traditional techniques the analysis would require a huge number of slides and a many years to analyze. Our approach combines multi-labeling (5 antigens can be simultaneously detected) and high-throughput imaging technologies. This strategy will allow us to carry out this analysis using an absolute minimum of precious NPOD slides in a precise manner.
-

Etienne Larger, Marc Diedisheim & Roberto Mallone
Université Paris Descartes
Beta-cell mass in Antibody-positive non-diabetic subjectsThe question of the B-cell mass of non-diabetic autoantibody-positive (Ab+) subjects is crucial to understand both common forms of type 1 diabetes and latent autoimmune diabetes (LADA). It is well established that the presence of GAD Abs is not sufficient to predict diabetes or insulin dependency. Knowing the B cell mass of Ab+ donors could be crucial to decipher the history of diabetes between the initial lesion, marked by the appearance of Abs, and the onset of diabetes. To this aim, we propose to use the insulin-stained virtual slides of the nPOD project to quantify the B cell mass of Ab+ subjects. We will first test whether the insulin-stained virtual slides of the nPOD database can be used for this analysis. To this end, we will compare the B cell mass, islet size distribution and B cell distribution throughout the pancreas in control donors of the nPOD database with published data of the literature. We will then compare the Ab+ subjects to control donors and patients with recent-onset, i. e. <3 years T1D.
-

Rudolph Leibel & Streamson Chua
Columbia University
Funded by: NIDDK
Identification of a gene regulating pancreatic beta cell replicationWe cloned a genetic modifier of T2D in obese mice. The gene, Ildr2, encodes a molecule that plays a role in both lipid metabolism and cellular responses to stress. Animals with lower levels of expression of this gene has reduced number of insulin producing cells in the pancreas, and, ultimately went on to develop mild diabetes. How the apparent functions of Ildr2 account for these effects is currently under study by us using genetically manipulated mice.
-
Rudolph Leibel, Wakae, Nao
Columbia University
Funded by: NIH
E-cadherin mediates developmental effects on the proliferation and the function of β-cells in the islet of LangerhansIn rodents and humans, the rate of beta cell proliferation declines rapidly after birth; formation of the islets of Langerhans begins perinatally and continues after birth. We tested whether increased levels of E-cadherin during islet formation mediate the decline in beta cell proliferation rate by contributing to a reduction of molecules that mediate cell replication. In vitro, a cultured beta cell line formed into clusters resembling an islet displayed increased E-cadherin but decreased nuclear β-catenin and cyclin D2 (cell division molecules) , and reduced rates of cell proliferation, compared with monolayers.
Reduction of E-cadherin increased cell proliferation and levels of cyclins D1 and D2. After birth, beta cells showed increased levels of E-cadherin, but decreased levels of D-cyclin, whereas islets of mice in which the E-cadherin gene had been knocked out showed increased levels of D-cyclins and nuclear β-catenin, as well as increased beta cell proliferation. These islets were significantly larger than those of control mice . Interestingly, these changes correlated with reduced insulin response to ambient glucose, both in vitro and in vivo. The findings support our hypothesis by indicating an important role of E-cadherin in the control of beta cell mass and function.
-

David Leslie
University of London
Risk of autoimmune disease and human self-antigen expressionWe have identified type 1 diabetes (T1D)-associated differential DNA methylated variable positions (T1D-MVPs) in CD14+ from peripheral blood mononuclear cells (PBMCs) from monozygotic (MZ) twins discordant for T1D (Rakyan V et al.; Plos Genetics 2011). We aim to determine the role of T1D-associated MVPs in the mechanisms that underpin immune dysfunction in T1D. We have therefore studied whether the same disease signature is detectable in T1D tissue compared with normal tissues relevant to the disease, namely: pancreatic lymph nodes (site of self-reactive T cells priming) compared with the spleen (as control). In addition we are examining the methylation state of T1D-MVPs in selected cells from human thymus and we intend to make these cells available to nPOD in due course.
-

Fred Levine
Sanford-Burnham Medical Research Institute
Funded by: CIRM
ORIGINAL: Beta-cell regeneration by transdifferentiation ADDENDUM: Assessment of Islet Cell Transdifferentiation in Type 1 DiabetesORIGINAL: We have been interested in the mechanisms by which beta-cells regenerate in diabetes. Our laboratory is interested in how beta -cells regenerate under normal and pathophysiological conditions, with the goal of developing new therapies for diabetes that result in an increased number of those cells. There are two possible mechanisms for endogenous regeneration: beta-cell replication and beta-cell neogenesis. In 2013, we published that potent small molecule HNF4a antagonists induced beta-cell replication in mice and rabbits. With respect to beta-cell neogenesis, we have developed a new model of pancreatic damage, combining pancreatic exocrine cell damage and chemical beta -cell ablation. In the model, high efficiency transdifferentiation of beta-cells to other islet cell types occurred. All of those experiments were done using mouse models. It is of interest to determine whether the findings in mice can be translated to humans. Thus, we propose to use samples from nPOD to examine for evidence of islet cell transdifferentiation.
ADDENDUM: The long term goal of this line of research is to induce the formation of new beta-cells by conversion of alpha-cells into beta-cells. We showed previously that alpha- to beta-cell conversion occurred in mice that had their beta-cells destroyed; similar to what happens in type 1 diabetes, but only when we also cause the animals to have pancreatitis, an inflammatory condition of the exocrine pancreas. We also found evidence of ongoing conversion of alpha-cells into beta-cells in humans with type 1 diabetes. In addition to alpha to beta-cell conversion, we found that beta-cells can convert into delta-cells, which make somatostatin in the islet. We studied a series of samples from nPOD, finding that there was progressive increase in the number of delta-cells with increasing duration of type 1 diabetes, even in older patients with many decades of type 1 diabetes. In some patients with a substantial number of preexisting beta-cells, we found extensive colocalization between insulin and somatostatin. Since alpha- to beta-cell transdifferentiation occurred only when there were few to no preexisting beta-cells, this suggested the possibility that beta-cells might convert directly into delta-cells. This would represent a paradigm shift in our understanding of the progression towards type 1 diabetes, as it would mean that beta-cells have a fate other than to die from autoimmune attack, which is to convert into delta-cells. The studies proposed here are designed to extend our previous observation to increased numbers of patients and to study unique patients with pancreas transplants who had recent onset recurrence of diabetes. We believe that by better understanding the process of islet cell transdifferentiation, it may be possible to develop the ability to induce the formation of new beta-cells in patients with type 1 diabetes.
-

Hua Lin & Domenico Accili
Forkhead BioTherapeutics, Inc.
FOXO1 inhibition-mediated reprogramming of b-like cells in gastrointestinal tissues from type 1 diabetic donorsGenetic inactivation of the nuclear transcription factor Forkhead box protein O1 (FOXO1) in mice in specific intestinal endocrine progenitor cells reprograms them into glucose-responsive insulin- producing (beta-like) cells, raising the promise that the gut can serve as an endogenous source for beta cell replacement. This phenomenon has also been demonstrated in vitro in cells derived from small and large intestinal tissues of normal human subjects and treated with FOXO1 inhibitors. Recent work in Dr. Accili’s lab identified a novel population of FOXO1-expressing cells in the glandular epithelium of the stomach in mice, which can be converted to beta-like cells upon FOXO1 genetic ablation. Whether the human stomach also harbors a FOXO1+ cell population that can be converted to beta-like cells is unknown. Furthermore, since type 1 diabetes (T1D) has been linked to gut epithelial dysfunction, questions remain as to whether the distribution of gut FOXO1+ cells in T1D resembles that in non-diabetics and whether the capability for these cells to convert into beta-like cells upon FOXO1 inhibition is preserved in T1D. Finally, whether the converted beta-like cells can survive ongoing autoimmunity in the T1D setting remains unknown.
The objectives of the proposed addendum is to leverage the expertise in Dr. Accili’s lab at Columbia University to characterize FOXO1 distribution in non-diabetic and T1D stomach tissues, use an in vitro system to determine the ability of FOXO1 inhibitors to convert cells in primary stomach cultures to beta-like cells, and examine the viability of the converted beta-like cells in an in vitro model of autoimmunity using autologous cytotoxic immune cells. The proposed work is expected to generate key in vitro proof-of-concept data to validate the stomach (in addition to the intestine) as a source of beta-like cell conversion by FOXO1 inhibitors and to inform an ongoing drug discovery program regarding the optimal absorption and distribution profiles for FOXO1 inhibitors as novel therapeutics for T1D.
-

Zhonghao Liu
NGM Biopharmaceuticals, Inc.
Target confirmation in human pancreasType 1 Diabetes (T1D) is caused by autoimmune-induced beta-cell destruction and successful beta-cell regeneration offers a potential cure to the disease. We have identified a beta-cell regenerative target that leads to the restoration of beta-cell mass in mouse models of beta-cell deficiency. We seek to confirm the observed cellular phenotype in human pancreatic samples.
-

Richard Lloyd & Joseph Petrosino
Baylor College of Medicine
Funded by: Baylor Diabetes Research Center
Virome and microbiome in T1D onsetT1D is recognized to result from both genetic and environmental factors. Chief among environmental factors that are strongly linked to T1D are Type B enteroviruses (HEV-B), yet the association of these viral triggers is not proven and many questions exist. Which of the 60 HEV-B serotypes trigger T1D, what type of infections do they cause in the pancreas, and how can they trigger autoimmunity are just a few. We are working within the nPOD-V, the virus working group, to attempt to amplify and isolate infectious viruses from nPOD samples, and to characterize the type of infections that occur in pancreatic cells. Upon amplification, samples are subjected to next generation deep sequencing to identify all viruses and microbes present in the samples in an unbiased manner. Samples are also examined using very sensitive qRT-PCR assays that detect single copies of enterovirus genomes. This work will provide the most complete picture of what infectious agents may be harbored in the pancreas of T1D patients, both before and after disease onset.
-
Richard Lloyd, Heikki Hyoty, Joseph Petrosino & Ivan Gerling
Baylor College of Medicine
Funded by: Breakthrough T1D and NIH-HIRN
Beta-cell stress signatures and infection in T1D isletsThis project will be the first to examine stress granule responses and the expression of long non coding RNAs in donor islet cells, a model beta cell line and the role of enterovirus infection in modulating these stress responses. This information will be used as a benchmark to examine nPOD tissues for these same stress responses, mobilization of innate immunity markers. The above experiments will also be performed in conjunction with analysis of the replication of terminally-deleted defective enteroviruses in the cell lines, and the stress responses they produce. This will inform secondary studies of actual nPOD tissues for the level of stress responses, key lncRNA expression profiles present. The nPOD tissues will also receive the most stringent analysis for virus capsid proteins and particularly viral RNA with single molecule sensitivity on a single cell basis that has yet been attempted.
-

Paolo Madeddu & Costanza Emanueli
University of Bristol, UK
Funded by: Wellcome Trust
Microangiopathy in diabetic bone marrowWe hypothesize that the alterations seen in diabetic BM are a major cause of impaired homeostasis in other vascular beds. Main objectives (1) to obtain a deeper insight into the cellular and vascular composition of diabetic BM using IHC, flow cytometry, and expressional studies (2) to determine whether BM microangiopathy is associated to poor metabolic control and increased susceptibility of cardiovascular complications. (adopted by nPOD staff)
-

Dorene Markel, Domenico Accili, Peter Arvan, Mark Atkinson, Jeffrey Bluestone, Matthias Hebrok, Kevan Herold, Chris Rhodes & Matthias von Herrath
Brehm Coalition, University of Michigan
Funded by: Breakthrough T1D
De-differentiation during progression of beta cell loss in type 1 diabetesThe overall goal of this proposal is to test the hypothesis that β-cell de-differentiation is an early stress-related response that may account for the early loss of β-cells in T1D. Furthermore, we believe that a small but significant population of long-term T1D patients might still have de-differentiated β-cells in their pancreatic islets, which creates hope for future re-differentiation paradigms. The ultimate goal is to address questions whose answers could lead to treatments that prevent β-cell loss and promote re-differentiation of progenitor-like cells into β-cells.
-

Mary Markiewicz
University of Kansas Medical Center
Funded by: NIH/NIAID, American Diabetes Association, & Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
NKG2D ligand expression in type 1 diabetes developmentRecent evidence derived from animal models of type 1 diabetes indicates that interaction of immune cells expressing the protein known as NKG2D with cells expressing other proteins, termed NKG2D ligands, plays an essential role in initiating diabetes development. In an effort to define more precisely the role these proteins play in type 1 diabetes progression, we developed novel mouse models. Based on results generated with these experimental models, we believe that expression of NKG2D ligands expressed on infiltrating immune cells within pancreatic islets is critical to the development of type 1 diabetes. Here we propose to confirm there is NKG2D ligand expression in the pancreas of type 1 diabetic donors.
-

Douglas Melton
Harvard University
Funded by: NIH
Generation of tools to distinguish human pancreatic cell populationsOur work aims to understand the defects were deficiencies in the pancreatic beta cells of diabetics. We use a number of molecular methods, including but not limited to single cell RNA sequencing, to characterize the molecular phenotypes of islet cells.
-

Sara Michie & Baohui Xu
Stanford University
Tissue-selective chemokines and adhesion molecules in human T1DMigration of lymphocytes from blood vessels into pancreatic lymph nodes (PanLNs) and pancreas is critical for the development of islet inflammation and beta cell destruction in type 1 diabetes (T1D). Adhesion of lymphocytes to the luminal surface of blood vessel endothelia in PanLNs and pancreas is an important step in this migration. However, the adhesion molecules that control migration of lymphocytes into human PanLNs and pancreas during the development of T1D are not well defined.
The goals of our project are to determine which endothelia adhesion molecules, and their lymphocyte receptors, are highly expressed in human PanLNs and pancreas during development of T1D, as compared to pancreas and PanLNs of controls (Aims 1 and 2). The physiologic importance of adhesion molecules that are expressed on lymphocytes and endothelia of PanLNs and pancreas during development of T1D will be determined using Stamper-Woodruff in vitro binding assays (Aims 3 and 4). Successful completion of these studies will help define the immunological mechanisms that are important for the development of islet inflammation and beta cell destruction in human T1D.
-

Anna Moore & Amol Kavishwar
Massachusetts General Hospital
Funded by: NIH
In vivo imaging of isletsThe aim of our study is to develop an imaging agent for in vivo determination of beta-cell mass in humans. Such an imaging agent will not only provide information on progression and management of diabetes but it will also help in monitoring an individual’s response to exercise, diet and anti-diabetic therapy. Unfortunately, there aren’t any proteins/epitopes that are known to be uniquely present on insulin producing beta cells that can be targeted for imaging. Unfortunately, imaging agents that are currently in use or are under development lack specificity. These imaging agents highlight other cell types as well on PET and MRI images making it difficult to estimate beta-cell mass.
In past we have shown the utility of an IgM antibody (IC2) for ex vivo determination of beta-cell mass. This antibody was around for several decades but lacked full characterization. We recently showed that this antibody binds to unique sphingomyelin-rich patches present on the surface of insulin producing beta-cells. We did not see such patches on any other cells or tissue that we tested. Further characterization using human diabetic samples from nPOD revealed that these unique sphingomyelin patches represent insulin-secretory status of beta-cells. These patches were present on beta-cells from pancreatic tissue from normal individuals. There were fewer patches on beta-cells from pancreatic tissues from diabetic individuals and they were completely absent from beta-cells from type-1 diabetic patients.
IgM is a very big molecule and owing to its propensity to activate complement and slow blood clearance, it is not an ideal molecule for use as an imaging agent. Currently, we are developing a shorter version of this affinity agent such that it can carry an imaging payload to beta-cells for in vivo imaging and estimation of beta-cell mass. In the future we plan to combine this affinity imaging agent with therapy delivery.
Interesting papers:
Diabetes. 2001 Oct;50(10):2231-6.
J Lipid Res. 2011 Sep;52(9):1660-71.
J Histochem Cytochem. 2013 Dec;61(12):910-9.
-

Conchi Mora
University of Lleida
Funded by: Spanish Ministry of Economy and Competitiveness
Differentially expressed genes in inflamed human isletsThe downregulation of cyclin D3 due to infiltration is causally related to beta cell dysfunction and beta cell-apoptosis in beta cells from NOD mice. We would like to confirm whether this is true in human beta cells. In order to validate cyclin D3 as a target for T1D therapy in humans, we need to evaluate the impact of inflammation on the expression levels of cyclin D3 in pancreatic beta cells from individuals at risk of developing T1D, newly diagnosed T1D patients and, non-medalist overtly diabetic patients, in comparison to control individuals, as an approach to study the disease in humans in relationship to cyclin D3 expression levels, as we have observed in NOD mice.
-

Grant Morahan, Patrick Concannon & Stephen Rich
University of Western Australia
Funded by: National Health and Medical Research Council of Australia and DRF
Genetics of nPODWe have found that people with type 1 diabetes fall into one of six subtypes that can be defined on the basis of the genetic variants they have. These subtypes differ with respect to various clinical features, including immunological traits and risk of diabetic complications.
In this proposal, we plan to use genetic information from the nPOD donors to define which T1D subtype they belonged to. This will allow analyses of findings from nPOD researchers to see if any of the features they are testing differ between the subtypes. This could be very important information for understanding T1D and future treatment of people with T1D.
In addition, we will set up a user interface that will allow nPOD researchers to test whether any of the information they have found from nPOD samples vary according to any of the genetic variants that have been tested.
-

Maggie Morris and Shannon Wallet
Eastern Virginia Medical School
Dissecting the role intestinal NK cells play in T1D pathogenesisThere is conflicting information regarding the role of natural killer (NK) cells in the development of Type 1 Diabetes (T1D). NK cells represent an important effector arm against viral infection, and mounting evidence suggests that viral infection plays a role in the development of T1D in at least a portion of patients. NK cells of diabetes prone NOD mice express high levels of the pro-inflammatory mediator 12-lipoxygenase (12-LO), and decreased levels of stimulatory receptors. NK cells of 12-LO deficient NOD mice, conversely, express significantly higher levels of stimulatory receptors. We previously found that 12-LO was upregulated in the pancreas of both T1D and T2D donors with insulin-containing islets. It is not clear how this 12-LO becomes upregulated, and whether or not additional sources of 12-LO may contribute to diabetes pathogenesis.
Therefore, we propose to study NK cells that reside in the duodenum of non-diabetic, T1D, and AAb+ donor samples. These cells will be characterized for their mRNA expression levels of anti-viral responses, as well as the expression of 12-LO and various key NK cell receptors. Further studies will assess the functionality of NK cells from different donors with respect to their ability to produce IFN-gamma and lyse target cells. Given the direct connection of the duodenum to the pancreas via the bile duct, there is a great potential for the immune cells in this area of the gut to influence the development of autoimmunity.
-

Jerry Nadler, Margaret Morris & Kaiwen Ma
Eastern Virginia Medical School
Funded by: Breakthrough T1D and NIH
12/15 Lipoxygenase expression in type 1 diabetes12/15-lipoxygenase (12/15-LO) is an enzyme responsible for the metabolism of arachidonic acid to pro-inflammatory responses by immune cells in a variety of tissues, including pancreatic islets and fat tissues. Our extensive research has shown that type 1-like diabetes development is significantly diminished in the absence of 12/15-lipoxygenase. By using 12/15-LO deficient C57BL/6 mice, we were able to show that these mice were protected from developing diabetes following multiple low-dose streptozotocin treatment. We then developed a non-obese diabetic (NOD) mouse line congenic for this deletion, and saw an extremely significant decrease in the rate of spontaneous diabetes as compared to wild-type NOD mice. We are moving forward to develop 12/15-LO inhibitors that might be used therapeutically. In order to maximize the productivity of these compounds, it would be extremely beneficial to garner information from type 1 and type 2 diabetic human samples. Namely, we are interested in testing the expression of 12/15-LO in human pancreas, lymph node, and spleen samples, so we can determine which cells produce this enzyme in the requested tissues.
-

Jerry Nadler, Margaret Morris & Julius Nyalwidhe
Eastern Virginia Medical School
Protein based biomarkers for type 1 diabetesWe have previously used Imaging Mass spectrometry (IMS) to identify Insulin within β-cells in the islets of Langerhans. The analysis utilized specially prepared pancreatic sections from the Network of Pancreatic Organ Donors with Diabetes (nPOD). In these studies, a comparative analysis of T1D MS spectra to those of Non-T1D revealed several uniquely expressed proteins between the two groups. In the current study, we propose to perform inter- and intra-sample qualitative and quantitative analyses to verify protein expression between normal and T1D tissue samples in an expanded subset of samples. We have developed methods that can distinguish proteins that are differentially present in T1D and non-diabetic samples. To this end, we have recently acquired a new instrument with a higher mass accuracy, resolution and sensitivity (Q-Exactive mass spectrometer, Thermo Fisher Scientific) on which we will perform LC-MS-MS analysis of additional samples in order to verify and validate the previously defined panel. Therefore, we propose to identify and develop quantitative assays to determine the utility of these molecules as potential diagnostic biomarkers for T1D by continuing our studies in additional pancreas samples.
-

Jerry Nepom
Benaroya Research Institute at Virginia Mason
Funded by: NIAID
Novel recombinant antibodies directed to diabetes-associated autoreactive T cell epitopesThe trimolecular complex composed of autoreactive T-cell receptor, MHC class II, and an autoantigenic peptide plays a central role in the activation of pathogenic islet-specific CD4+ T cells in type 1 diabetes (T1D). We isolated and characterized novel antibodies against autoreactive T-cell epitopes associated with T1D. Our antibodies mimic the specificity of the T cell receptor (TCR), while binding MHC class II/peptide complexes in an autoantigen peptide-specific, MHC-restricted manner. The isolated TCR-like antibodies were directed against the minimal T cell epitope GAD-555-567 in the context of the HLA-DR4-diabetic-associated molecule. A representative high-affinity TCR-like antibody clone (G3H8) enabled the detection of intra- and extra-cellular DR4/GAD-555–567 complexes in APC. I561M single mutation at the central position (P5) of the GAD-555-567 peptide abolished the binding of G3H8 to the DR4/GAD complex, demonstrating its high fine TCR-like specificity. The G3H8 TCR-like antibody significantly inhibited GAD-555-567 specific, DR4-restricted T cell response in vitro and in vivo in HLA-DR4 transgenic mice.
Our findings constitute a proof of concept for the utility of TCR-like antibodies as antigen-specific immunomodulation agents for regulating pathogenic T-cells and suggest that TCR-like antibodies targeting autoreactive MHC class II epitopes are valuable research tools, which enable studies related to antigen presentation, as well as novel therapeutic agents, that may be used to modulate autoimmune disorders such as T1D.
Interpretation
We have demonstrated that G3H8 can be used to detect and visualize GAD presentation by APC. The TCR-like fine specificity and requirement for both GAD-555–567 peptide and the HLA-DR4 molecule for G3H8 recognition were verified. Moreover, we have identified the P5 position in the center of the peptide to be essential for G3H8 binding of the DR4/GAD complex. This position was found to play a critical role in T cell recognition [24], and thus the overlap in P5 dependency between the TCR and our G3H8 TCRL antibody implies that our antibody has fine specificity and TCR-like characteristics. Once the fine specificity of G3H8 was determined, we used it to detect naturally processed GAD epitopes. G3H8 stained specific MHC class II
complexes derived from processing of the whole GAD antigen by GAD-transfected Priess cells. The lack of extracellular staining by G3H8 suggests that most of the DR4/GAD complexes in these cells were located inside the cells, whereas the number of complexes presented on the cell surface was below the detection threshold of G3H8. Our failure to stain nPOD pancreata is likely due to this same issue, and in addition we had background issues that were never resolved. We concluded that use of this novel reagent needs a substantial amount of the epitope on the APC for detection (only seen when peptide was added to cells or after an immunization in mice). -

Janelle Noble
Children’s Hospital Oakland Research Institute
High resolution HLA typingThe Noble lab produces HLA genotyping for all nPOD samples.
-
Janelle Noble
Children's Hospital Oakland Research Institute
Assessing heterogeneity of DR3 haplotype risk in type 1 diabetesWe have discovered a new DNA variation that we think may affect T1D risk in individuals carrying a particular type of HLA-DRB1 gene. We aim to look at particular nPOD samples with that HLA genotype to see if they contain our newly discovered variant. If so, we will be able to design a future project to address the potential function of the variant. This is a rapid, exploratory experiment.
-

Hervé Perron & Virginie Vidal
Geneuro, Switzerland
Evaluation of HERV-W Envelope antigen expression in Pancreas and serum from patients with Type 1 Diabetes.Endogenous retroviruses are known to represent 8% of the Human genome. HERV-W family retains elements expressing an envelope protein (Env), which activates a pro-inflammatory and autoimmune cascade through interaction with Toll-Like receptor 4 (TLR4). This Env protein was evidenced in brain lesions, sera and circulating mononuclear cells of patients with Multiple Sclerosis (MS)1. Thus, all the background science from the past two decades that has brought independent confirmations and reproductions of an involvement of HERV-W in MS 2 convinced us to further study its association with other “autoimmune diseases”. If no association was found when testing series of patients with rheumatoid arthritis3 or systemic lupus1, about one-third of sera from patients with T1D revealed positive for the HERV-W Envelope antigen. We therefore further explored whether this immunopathogenic endogenous protein could be expressed in pancreas from T1D versus controls, which revealed positive in about 40% of first pilot series, and are now preparing to extend these analyses to larger number of serum, PBMC and pancreas samples. In parallel, we are developing an HERV-W-Env induce mouse model of diabetes for pre-clinical studies with an anti-Env neutralizing humanized antibody.
References:
1. Perron H, Germi R, Bernard C, et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult Scler. 2012; 18: 1721-36.
2. Dolei A and Perron H. The multiple sclerosis-associated retrovirus and its HERV-W endogenous family: a biological interface between virology, genetics, and immunology in human physiology and disease. J Neurovirol. 2009; 15: 4-13.
3. Gaudin P, Ijaz S, Tuke PW, et al. Infrequency of detection of particle-associated MSRV/HERV-W RNA in the synovial fluid of patients with rheumatoid arthritis. Rheumatology (Oxford). 2000; 39: 950-4.
-

Thomas Pieber
Medical University of Graz
Linking IHC immune profiles of duodenum and pancreas in T1DOur project “Linking IHC profiles of duodenum and pancreas in T1D” aims to intensify our understanding of how the “gut immune system” contributes to the development of type 1 diabetes.
Previous studies have shown that factors like the gut microbiome, gut permeability and the mucosal structure play a role in T1D pathogenesis. Studies are now more and more focusing on intestinal immune cells and their contribution to the onset and disease course of T1D. Regulatory T cells (Tregs) are of special interest in this regard, since they are capable of suppressing auto-reactive effector T cells, and thereby prevent autoimmune reactions. The role of intestinal Tregs in T1D development has been described controversially. While some papers report of increased numbers of Tregs in the intestine of T1D patients, other publications find no difference in Treg numbers between patients and healthy subjects. With our new immunohistochemical method, which allows us the identification of CD4+CD25+FoxP3+ putative regulatory T cells with a specificity that has not been reached before, we hope to shed light on regulatory T cell profiles of duodenum and pancreas in parallel in patients at different disease stages compared to healthy subjects. Additionally we aim to investigate CD8+ and CD4+ Th17 effector T cells in the same tissues in order to get comprehensive information on regulatory and effector T cell subsets. Furthermore, we will investigate T cell function by using single-cell analysis of positively stained T cells regarding their expression profiles and methylation status.
Thus, using the precious nPOD samples, we hope to identify immunological processes in the duodenum and pancreas that play a crucial role in the development and pathogenesis of type 1 diabetes.
-

Lorenzo Piemonti & Ilaria Capua
San Raffaele Scientific Institute, Italy
Funded by: Italian Ministry of Health
Role of influenza viruses in the etiopathogenesis of diabetesThe rapid worldwide incidence increase suggests a major role for environmental factors in the aetiology of type 1 diabetes (T1D). According to cross-sectional and prospective studies on T1D patients and/or prediabetic individuals, virus infections may be one of these. Influenza viruses are known to cause severe pancreatic damage in birds and in some mammals, and we have preliminary strong evidence that these virus growth in primary pancreatic islet (human and mouse) cultures “in vitro”. The objective of our project is to demonstrate that influenza viruses may play a role in the etiopathogenesis of T1D. Due to the interdisciplinary nature of this study, two different Research Institutes will be involved: Istituto Zooprofilattico Sperimentale delle Venezie (I Capua, PI) for the influenza virus expertise and the San Raffaele Diabetes Research Institute (L Piemonti, CoPI) for the diabetes expertise.
The overall goal of this research is to demonstrate that influenza virus infection may be involved in the etiopathogenesis of diabetes.
Specifically we will aim to:
1) Test “in vitro” the ability of influenza viruses to infect, replicate, and damage beta cell, pancreatic islet and endocrine cell
2) Test “in vivo” (mouse) the ability of influenza viruses to induce beta cell damage and/or alterations of glucose metabolism
3) Look for direct and/or indirect evidences of correlation between influenza viruses infection and autoimmunity/diabetes onset in humans
For the aim 3 we are testing the presence of influenza virus in pancreas at disease onset in T1D patients. For this purpose tissue samples are asked to the Network for Pancreatic Donors with Diabetes (nPOD). In situ hybridization will be performed to visualize viral RNA localized within cells using the Quantigene ViewRNA technique based on branched DNA signal amplification technology. Moreover the presence of influenza virus will be eventually confirmed by digital PCR and immunohystiochemistry
-

Massimo Pietropaolo & Michael Morran
Baylor College of Medicine
Funded by: NIDDK
Identification of T1D-specific Fab fragments of IA-2 dominant conformational epitopeOne of the primary characteristics of Type 1 Diabetes (T1D) is the presence of autoantibodies directed to self-antigens expressed in the pancreatic islets, specifically those associated with the insulin secretory machinery. Although T1D is well characterized as a T-cell mediated disease, a mechanistic role of islet autoantibodies generated during the progression of T1D have not been elucidated. The tyrosine phosphatase-like protein (IA-2) is a major neuroendocrine autoantigen associated with the progression of T1D. We previously identified a new candidate biomarker within the extracellular domain of IA-2 (IA-2ec) in humans, which is associated to rapid acceleration of T1D onset compared to conventional IA-2 antibody biomarkers of T1D. Little is known about the clonality and the V region structures of IA-2 autoantibodies, an important consideration given their diagnostic and predictive value. Thus, our objective is to perform de novo sequencing to determine the clonality and V region structures of human autoantibodies directed against the islet autoantigen IA-2. This method can be applied to multiple serum samples and has been validated by the discovery of a clonotypic autoantibody specific for an immunodominant determinant of target antigens of a number of autoimmune diseases including systemic lupus erythematosus (SLE) rheumatoid arthritis (RA). In particular, we aim to characterize the IA-2 autoantibody sequence in the sera of the well-characterized population of T1D patients of the nPOD network in an effort to determine if these autoantibodies express a highly restricted B-cell clonotype repertoire which may be shared (public) across different disease phenotypes. We will then identify unique V region peptides to interrogate human sera for IA-2 by a targeted mass spectrometry (MS) protocol. Mass spectrometry-based detection of specific autoantibody motifs may provide a powerful new tool for analysis of humoral autoimmunity in T1D.
-

Jon Piganelli & Eddie James
University of Pittsburgh
Funded by: Breakthrough T1D
Disruption of tolerance by ER-stress in type 1 diabetesType 1 diabetes (T1D) is an autoimmune disease in which insulin-producing islet cells are targeted and destroyed by the immune system, especially by T cells. The events that lead to immune recognition of islet proteins in people genetically predisposed to autoimmunity are poorly understood. Many of the environmental triggers associated with T1D including viral infection, exposure to chemicals or free radicals, and dynamic glucose sensing, however, cause cellular stress within islets. In addition, the natural function of islet cells (to produce and release insulin) makes these cells especially susceptible to stress. The cells respond to stress by activating a program that protects them from death, but this results in mishandling of proteins leading to changes in there normal structure. In healthy individuals, this mishandling of proteins is not a problem; however, in individuals prone to autoimmunity, these proteins and the cells that make them may be targeted by the immune system. We hypothesize that the environmentally related events leading to this cellular stress in islet cells causes abnormal modification of proteins which in genetically predisposed patients, leads to abnormal immune system targeting causing the destruction of the islets. We have tested this hypothesis using a model system of T cell recognition of islets, in particular an islet protein called chromogranin A (CHgA) in a well established mouse model of T1D, demonstrating that T cells respond much more strongly to islets exposed to environmental triggers of cellular stress. These data support the hypothesis that cellular stress changes the proteins in the islet, and these changes facilitate the recognition of islets by T cells. We are now proposing to translate these findings further using human islet-reactive T cells, human islets and a humanized mouse model of T1D. The overarching goal of these studies is to determine in the humanized T1D animal model if the same modifications are demonstrated under conditions of cellular stress, and to determine how these modifications lead to the recognition of islets by the immune system. These studies are important because understanding how islet proteins come to be recognized by T cells will reveal targets for therapeutic intervention to prevent incorrect modification of islet proteins, protect islets from the immune system, and ultimately prevent T1D.
-

Andrew Pospisilik & Tess Tsai-Hsiu Lu
Max Planck Institute of Immunobiology and Epigenetics
Epigenetic regulation of beta-cell function and identityDiabetes is a disorder of complex interaction between genetic susceptibility and environmental perturbation. Various epigenetic models, such as intra-uterine growth retardation, have been shown to contribute to pancreatic beta-cell dysfunction. More recently, beta-cell dedifferentiation has been discovered as a mechanism for beta-cell dysfunction in diabetic mouse model. The project aim to understand the role of classic epigenetic modules in regulating beta-cell identity and function in human context. We’ll survey changes in histone modifications across human pancreas samples over various age groups and diabetic conditions. Understanding how histone modification changes with age and disease state in human will enable the research community to identity factors that underlie aging related decline in beta-cell function and identity.
-
Alberto Pugliese & Francesco Vendrame
Diabetes Research Institute, University of Miami
Funded by: Assessment of memory T cells in the insulitis lesion
Assessment of memory T cells in the insulitis lesionThere is growing evidence that memory T cells are associated with a variety of autoimmune conditions, including type 1 diabetes (T1D). While autoreactive T cells are detected in both patients and healthy control subjects, CD45RO+ memory autoreactive T cells are usually found in patients but not in controls. Our own studies in several pancreas transplant recipients with T1D recurrence have shown that autoreactive memory T cells can be found in the circulation and pancreas transplant lymph nodes. Moreover, the examination of pancreas transplant biopsies from two of such patients shows that most (~80%) of the islet infiltrating T cells have a memory phenotype. Based on this findings we hypothesize that memory cells should be present in the islet infiltrates of nPOD patients. Specifically, we plan to study pancreatic tissue obtained from patients with proven insulits for the presence of the T cells bearing the memory marker CD45RO. It is plausible that the frequency of the memory cells may be related to disease duration, and perhaps this could be higher in the lesions of patients with longer disease duration who still have insulitis. It is also possible that there will be a difference compared to pancreas transplant recipients with recurrent disease. Understanding whether memory T cells represent a significant component of islet infiltrating T cells is relevant to a more complete characterization of the insulitis. A recent clinical trial in new onset patients has tested Alefacept, an anti-memory T cell drug. Demonstrating memory T cells in the lesion would provide further rationale for regimens that target memory T cells, with Alefacept or with new drugs that have similar effects. Furthermore, if memory cells are demonstrated, we can propose additional work to better define the memory phenotype by staining for additional markers.
-

Sasanka Ramanadham
University of Alabama-Birmingham
Funded by: NIDDK, UAB Comprehensive Diabetes Center and Iacocca Family Foundation
Role of Calcium-Independent Phospholipase A2β (iPLA2β) on β-Cell ApoptosisBeta-cell apoptosis contributes to loss of β-cells and decreases in β-cell function in both types 1 and 2diabetes mellitus. It is therefore important to understand the mechanisms underlying β -cell apoptosis if this process is to be prevented or delayed. Our hypothesis is that the group VIA Ca2+-independent phospholipase A2 (iPLA2 β) participates in β -cell apoptosis. We observed that (a) ER stress, proinflammatory cytokines, and prolonged hyperglycemia promote expression and activity of iPLA2 β inislets, (b) iPLA2 β activation increases ceramide generation via neutral sphingomyelinase (NSMase)- catalyzed hydrolysis of sphingomyelins triggering the mitochondrial apoptotic pathway and β –cell apoptosis, (c) these outcomes are suppressed by inhibition of iPLA2 β or NSMase, (d) iPLA2 β –null islets are less and iPLA2 β -transgenic (Tg) islets more sensitive to ER stress, (e) NSMase expression is unaffected in iPLA2 β -null islets and amplified in iPLA2 β -Tg islets, and (f) β -cell iPLA2 β and NSMase messages are higher in the Akita mouse model of spontaneous β -cell ER stress that leads to diabetes. We also find that iPLA2 β participates in human islet β -cell apoptosis induced by hyperglycemia and cytokines and that islet iPLA2 β and NSMase messages are elevated in prediabetic NOD mice. Further, we recently found evidence for inhibition of iPLA2 β leading to decreased incidence of T1D in diabetes-prone NOD mice. These findings strengthen our hypothesis thatiPLA2 β participates in the onset and progression of T1D. We propose to validate our hypothesis in human T1D by correlating expression levels of iPLA2 β with indices of beta-cell apoptosis in pancreas samples obtained from control and diabetic donor subjects.
-

Shiva Reddy
University of Auckland, New Zealand
ORIGINAL: Immunohistochemical identification of molecular markers of oxidative and nitrosative stress in β-cells during the early stages of human type 1 diabetes mellitus ADDENDUM: Beta cell stress as an early preceding phase of autoimmune type 1 diabetes (T1D): Immunohistochemical assessment of markers of early beta cell vulnerability in newly-diagnosed and long-term diabetic casesORIGINAL: This research seeks immunohistochemical evidence of beta cell oxidative and nitrosative stress during various stages of T1D and whether such stressors are also present preceding and during the early onset of the disease. Antibodies to nitrotyrosine, a marker of peroxynitrite-mediated cell damage and to 8-hydroxy-2-deoxyguanosine (a marker of DNA oxidation) are being employed in triple-label immunohistochemistry to determine if such processes are activated within the beta cell. Combined immunohistochemistry and histochemistry are being applied to correlate the immunolabelling of such deleterious markers with the extent and spatial distribution of leukocytic infiltrates within the islets and with beta cell co-localization. Studies to date have shown weak to moderate immunolabelling of nitrotyrosine restricted to a majority of the remaining beta cells from 5 T1D cases, being also present in islet beta cells with minimum insulitis. Nitrotyrosine was absent in glucagon cells and the islet leukocytes. It was absent or weakly expressed in islet cells of non-diabetic donors (2 cases). DNA oxidation was observed within islet endocrine cells but was not restricted to beta cells from T1D cases.
The expression of nitrotyrosine and DNA oxidation are being extended to non-diabetic cases with single or two autoantibodies. In line with previous in vitro studies with human islets in culture, the current research is also exploring the possible activation of the pro-inflammatory cytokine-nitric oxide axis within the islets of T1D cases, likely under-expression of anti-oxidant enzymes and whether stressors within the pre-diabetic beta cell lead to beta cell apoptosis and attract the early immune response.
Protocol development, and search for suitable primary antibodies and optimization studies are in progress for the immunohistochemical localization of interleukin-1β, inducible nitric oxide synthase and interferon-α.
ADDENDUM: Despite considerable efforts directed at finding a cure for type 1 diabetes (T1D), the molecular processes that initiate and perpetuate beta cell destruction and how and why beta cells become targets of immune destruction are poorly understood.This knowledge gap and the realization that the underlying processes of T1D and the age of onset of T1D are heterogeneous have compounded progress in finding a cure.In addition, evidence of beta cell dysfunction is present well before clinical onset.
Beta cells have high rates of metabolism, protein synthesis and secrete insulin continuously in a pulsatile fashion and with increased output in response to meals. We propose that the highly metabolic beta cells are more susceptible to environmentally-induced insults, that oxidant-induced beta cell stress, leading to generation of abnormal cellular proteins due to their intrinsic inability to clear excess free radicals. The resulting abnormal proteins compromise beta cell function and act as antigenic targets of immune-mediated destruction.
Environmental factors such as certain viruses or toxins can also lead to excess and uncontrolled generation of reactive oxygen species (ROS) in beta cells, resulting in stress.Thus, sustained beta cell stress may be a key determinant of beta cell damage. In this research, we wish to demonstrate if the remaining beta cells from cases with new-onset T1D and those with longer duration of disease have low levels of three key ROS clearing enzymes, and express detectable levels of interferon-α (IFN-α), a marker of virally-infected beta cells and which my contribute to oxidative stress.The endoplasmic reticulum (ER), a vital subcellular organelle, is responsible for correctly processing and folding of nascent proteins and is exquisitely sensitive to imbalances in oxidative stress. We also wish to examine if oxidative stress disturbs the redox balance and promotes stress within the ER.
Our studies will provide key insights on why beta cells become targets of self-immunity and may pave the way for the development of future therapies aimed at alleviating beta cell stress in the early stages of type 1 diabetes and even prior to its onset and thus, prevent this serious disorder.
-

Christopher Rhodes
University of Chicago
“Empty Beta Cells” in type 1 diabetesDuring the pathogenesis of Diabetes Mellitus, whether it be type 1 or 2 diabetes, islet cell heterogeneity becomes apparent. This is most commonly seen as a marked variability between the insulin content of some ß-cells versus others. Indeed, some ß-cells have such low insulin content that they appear to be empty. As such, in using insulin as a marker to estimate ß-cell numbers (as commonly done), one could be under-estimating the actual numbers of ß-cells presented since ‘empty ß-cells’ would not be accounted for. The concept of ‘empty ß-cells’ in the pancreas of diabetic patients could alter current thinking on ß-cell growth and regeneration. And, of course, if ß-cells are present in significant numbers, then it may become an issue of suitably refilling these cells with insulin rather than replacing/regenerating them.
We are interested in further examining the basis of ß-cell heterogeneity and ‘empty ß-cells’. Is this symptomatic of ß-cell dedifferentiation, a degranulated ‘hard working’ ß-cell and/or a stressed ß-cell approaching its end. We have used the transcription factor MafA as an alternative specific marker of adult pancreatic ß-cells, where a MafA+/Insulin- cell would indicate an ‘empty ß-cell’. But MafA is also susceptible to oxidation under oxidative stress conditions (e.g. hyperglycemia) and thus we are interested in examining other markers of ß-cell specificity to complement this analysis. We are also interested in examining adaptive plasticity of ß-cells and what mechanism guides the expansion of the rough endoplasmic reticulum, Golgi apparatus and trans-Golgi network reflective of upregulated insulin production to meet the increased demand in diabetes. Finally, we suspect that oxidative stress is a prime contributor to ß-cell destruction in the pathogenesis of diabetes and want to examine if an ‘empty ß-cell’ marks susceptibility to such stress and/or ß-cell very close to an apoptotic state.
-

Stephen Rich
University of Virginia
Funded by: NIDDK
Immunochip AssaysnPOD participated in the ImmunoChip consortium, that was established to design a cost effective genotyping array in order to fine map well established GWAS reported risk loci in immunologically related human diseases, including type 1 diabetes.
-

Daniel Rios
Pandion Therapeutics
Longitudinal Determination of Islet Specific protein expression in T1DPandion Therapeutics is a biotechnology company developing bifunctional protein therapeutics to achieve localized immunomodulation at the site of disease for durable, tissue-specific treatment of patients with autoimmune and inflammatory disease and organ transplant. Our localized approach represents a significant change from the systemic immunosuppression of conventional medicines. Through tissue-specific therapeutic targeting, Pandion’s approach has the potential to more effectively induce and sustain response and remission in patients with autoimmune and inflammatory conditions. In collaboration with the Breakthrough T1D and Astellas Pharmaceuticals, we propose to develop new therapeutics to treat patients with T1D.
Our bifunctional therapeutics, by definition, have two functional activities – targeting and immunomodulation. Given the progressive nature of T1D it is critical to understand the protein expression on a cell-by-cell basis during the progression of T1D in order to develop tissue targeting bifunctional therapeutics. We have currently identified several potential candidate tether molecules for pancreatic targeting. However, currently it is unknown if the expression (total level, different isoforms, post-translational modifications) of these tissue-specific molecules changes during the progression of T1D. Furthermore, in order to enable FIH studies it is of the utmost importance that we demonstrate that our newly generated bifunctional proteins are able to localize to the intended tissue site, in this case pancreatic islets, prior to dosing in patients with T1D. This project seeks to understand the temporal regulation of pancreas specific proteins during the progression of T1D and confirm specific binding of our therapeutic bifunctional molecules. We hope to gain a better understanding of how these proteins are regulated, if at all, over-time to determine if these targets are suitable for prophylactic or therapeutic tissue-specific immunomodulation.
-

Bart Roep
Leiden University, Netherlands
Detection of islet autoreactive CD8 T-cells in insulitis versus peripheryTo determine the specificities and frequencies of islet autoreactive CD8 T-cells in pancreas draining lymph nodes, control lymph nodes, spleen and/or peripheral blood from diabetic and non-diabetic organ donors, we used the Diab-Q-kit, a nanotechnology that allows the direct detection of circulating autoreactive CD8 T-cells against an array of islet-epitopes simultaneously, reducing the needs of patient resources. The detection of these T-cells is based on the T-cell receptor (TCR) specificity for its peptide-HLA ligand. We produced HLA-monomers for an array of islet epitopes presented in the context of HLA -A2, -A3 and -B7. This allowed expanding the number of patients that could be analyzed considerably. Moreover, to characterize the memory phenotype of the autoreactive T-cells, we validated and upgraded the Diab-Q-kit through the inclusion of cell surface markers to distinguish naïve, effector memory and central memory T-cells. From the nPOD repository we selected a total of 29 cases to be analysed for this study from which we could obtain cell suspensions from blood and pancreas draining lymph nodes, control lymph nodes and/ or spleen.
Upon thawing of the samples we unexpectedly observed that far fewer viable cells could be recovered from the majority of the samples than the number reported to be frozen on the vial. For some cases, we also counted the number of dead cells in each vial. We found that the percentage of dead cells was very high in some cases, but not all. As an internal control, we always included samples from healthy controls collected and cryopreserved in our laboratory, which showed very high viability after thawing (>90%), indicating that the thawing procedure was correctly performed.
Similar issues on viability and recovery have since been reported by other partners in nPOD. Importantly, the quality control to detect any issues was critical to identify these problems and potential causes. For instance, issues with viability and recovery were determined immediately after thawing, while FACS analyses performed hours after thawing of the samples would have suggested excellent viability outcome, even in cases that QC earlier on had identified poor recovery and viability.
The consequences of low viability and recovery may differ depending on the type of analysis per project (functional assays vs. mRNA) and incentive (cloning T-cells may still be possible, but may not be representative of the total tissue sample). Such issues have been evaluated in depth and at length between the central facility and participants of nPOD. Indeed, the nPOD central facility was indispensable in identifying important correlations between recovery, viability and donor characteristics that may differ between tissue source of the leukocytes and that need not be independent co-variates ( PBMC: demographics, cause of death [anoxia vs > CV/stroke], disease [T2D/Ab+>T1D], donor age [o>y]; splenocytes: donor age [y>o], BMI [low>hi], transmission time [short>long]), underscoring that the thawing procedure was not the primary parameter of poor outcome and implying that intrinsic features of the biosamples determines quality and reward.
Despite lower cell recovery than expected, we nevertheless proceeded performing the Diab-Q-kit analysis on all samples, to take maximal knowledge, benefit and reward from the precious nPOD material. Unfortunately, for more than half of the samples no reliable data could be obtained, due to the low number of CD8 T-cells present in each sample. This precluded reliable comparisons in frequencies and specificities of islet-autoreactive CD8 T-cells in the individual organs. The results from this study have been discussed at length with nPOD, and it has helped the nPOD consortium to decide on the feasibility of projects where viable cell suspensions are needed for analysis and functional assays. Measures have been undertaken to adjust allocations of samples on basis of this important new insight.
-
Tamás Röszer
University of Ulm
Funded by: European Union, German Research Fund
Hormonal regulation of adipose tissue macrophage functions in diabetesOur research is focused on the understanding of hormonal control of adipose tissue macrophage (ATM) function. ATMs play key roles in diabetes development: when they adopt an inflammatory activation state, they inhibit insulin signaling, cause systemic insulin resistance, exacerbate autoimmunity and pancreatic beta cell destruction. On the contrary, when ATMs undergo a so-called alternative activation, they support metabolic performance. Understanding the hormonal signals which are decisive for these activation states is thus key for treating diabetes. The current projects in our laboratory study the role of pancreatic hormones on ATM function.
-

Srinath Sanda & Rebecca Hull
University of California, San Francisco
Funded by: NIH
IL-1beta expression in cystic fibrosis diabetesCystic fibrosis related diabetes is a major morbidity for patients with cystic fibrosis and negatively impacts mortality. The mechanistic basis for this form of diabetes is not well understood. In this project we are analyzing the hypothesis that islet inflammation leads to islet amyloid deposition and beta cell failure in patients with CFRD. Samples from the nPOD repository are being stained for both amyloid and interleukin 1 beta. In the future we will look at additional inflammatory mediators in the islets.
-

Maike Sander
Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Germany
Preventing beta cell dedifferentiation in type 1 diabetesRecent evidence suggests that beta cell dedifferentiation is a key early mechanism that underlies the pathogenesis of type 2 diabetes (T2D) [1]. This includes the loss of transcriptional regulators of beta cell function as well as other critical components of beta cell function. The study further suggests that beta cell dedifferentiation and acquisition of progenitor markers precedes beta cell death in T2D. Whether the same sequence of events and loss of factors occurs during progression of type 1 diabetes (T1D) is currently unknown. Interestingly, the analysis of pancreata from T1D individuals suggests that T1D patients retain insulin-producing cells even years after the onset of disease [2]. However, preliminary, still unpublished evidence suggests that these remaining beta cells have impaired health and have lost expression of some critical beta cells genes (Mark Aktinson, ADA meeting, June 2012). This observation raises the question of how function and mass of these remaining beta cells could be improved. We hypothesize that similar factors as lost in T2D are also lost in T1D and result in impaired function of remaining beta cells.
The overall goal of this proposal is to identify pathways that can prevent and/or reverse beta cell dedifferentiation in type 1 diabetes. The strategy is to define upstream pathways that maintain the expression of critical beta cell differentiation factors. It is predicted that sustained expression of these differentiation factors will restore beta cell function and mass in type 1 diabetes.
-

Suparna Sarkar & Dirk Homann
Mount Sinai Hospital
Funded by: Breakthrough T1D and NIDDK
Pancreatic expression of chemokines in human type 1 diabetesHuman chemokine superfamily of molecules mediate the recruitment of leukocytes to sites of inflammation. Many chemokines and chemokine receptors have emerged as key contributors and regulators of autoimmune disorders, including Type 1 Diabetes and may serve as potential surrogate biomarkers of inflammation or possible drug targets. More than one-half of all human and rodent chemokines have been implicated in the pathogenesis or complications of Type 1 diabetes. Most of the previously published work however is limited to the genetic association studies and chemokine and their receptor analyses in the peripheral blood. To delineate the complete spectrum of chemokine mRNA species transcribed by human islets in response to inflammatory stimuli, we used an established in vitro culture system in which islets purified from healthy organ donors were cultured in the absence versus the presence of IL-1β, TNFα, and IFNγ and subsequently subjected them to gene expression microarray analysis. Based on our microarray data and in collaboration with Homann laboratory, we study the expression and localization of chemokines in pancreatic tissue sections obtained through the Network for Pancreatic Organ Donors with Diabetes (nPOD) and included healthy control subjects.
-

Nora Sarvetnick & Kamiya Mehla
University of Nebraska Medical Center
Funded by: NIH
Analysis and comparison of IL-18, IL-18R, CCL19 and CCL21 expression in T1D, T2D, autoantibody positive and control samplesType 1 diabetes (T1D) has devastating effects on the health and quality of life of those affected. Several studies have associated the proinflammatory cytokine IL‐18 with an increased risk of T1D. Significantly higher IL-18 cytokine levels are seen in individuals at high risk for developing disease, increased serum levels of IL‐18 have been observed in T1D, and the presence of multiple islet autoantibodies in new‐onset diabetics was found to be associated with increased serum levels of IL‐18. Furthermore, our recent investigations demonstrate a role for IL‐18 in expanding the pool of islet‐destructive T cells during pre‐diabetes. Taking the elevated serum levels of IL-18 in T1D and enhanced survival properties of IL-18 receptor (IL-18R)-expressing CD8 T cells into account, there might be a connecting link between IL-18R+ CD8 T cells and T1D pathogenesis. Hence, in the present study we propose that IL-18R+ CD8 T cells may underlie the direct or indirect islet destruction associated with T1D. Furthermore, IL-18 expression has been linked to the C-C chemokine receptor type 7 (CCR7). Specifically, IL-18 has been shown to induce expression of CCR7, and IL-18-activated cells can stimulate the production of the CCR7 ligand CCL19. Based on this data and our preliminary data showing an increase in proportion of a transitional CD4 T cell population expressing CCR7 in new-onset T1D patients, we postulate that the CCR7 ligands CCL19 and CCL21 will be overexpressed in the pancreas and spleen of T1D patients when compared to control, autoantibody positive, and T2D patients. Overall, the goal of our studies is to fully understand the role of IL-18R+ CD8 T cells and associated cytokines and soluble factors in T1D pathogenesis. With the aid of nPOD, human spleen and pancreatic tissue specimens will be utilized for immunohistochemical studies to determine islet infiltration of IL-18R+ CD8 T cells, as well as differential expression of cytokines and soluble factors important for the development of T1D.
-
Nora Sarvetnick
University of Nebraska Medical Center
Funded by: NIH/ Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
Pancreatic antibacterial responses in T1DThe etiology of autoimmune diabetes is not known. However, the disease is accompanied by varying amounts of immune activation and inflammation. Although genetics clearly plays a role in determining who gets the disease, we do not know the instigating events. There has been a lot of investigation into the potential role of virus infections in triggering disease. However, there are no direct associations known between virus and disease in humans.
Recently, there has been increased interest in issues such as intestinal permeability and the microbiome in diabetes patients. One theory is that the leaky gut in T1D patients allows the penetration of bacteria and/or their products into the body, causing immune stimulation. When such products arrive into the pancreas after draining from the ducts, they cause changes in gene expression and inflammation. One set of factors that would get induced is bactericidal proteins, whose expression or subcellular localization is affected by exposure to bacterial products.
We have studied the expression of bactericidal proteins in circulation in new onset T1D patients. These studies indicated that there were coordinately high levels of expression in circulation in T1D patients less that five years after disease onset. These results suggest the upregulation of anti-bacterial responses in T1D patients. In this project, we will determine whether bactericidal factors are upregulated in the pancreas in T1D patients.
This project is important because it will tell us whether the disease may be mediated by the release of bacteria or their products into the pancreas, which would cause immune activation and inflammation. Several different bactericidal response elements will be investigated, each reflecting exposure to different bactericidal products including DNA, cell membrane constituents, and pigment molecules, an each providing an indication of the type of exposure the pancreas has encountered.
-

Bruce Seligmann, Klaus Pechhold & Horacio Rilo
BioSpyder Technologies, Inc.
In situ gene profiling and identification of T1D disease mechanismsThis CBDS program will establish an in situ RASL-Seq Islet Study Facility that will validate a novel in situ RASL-Seq platform for diabetes research using FFPE tissue from normal and Type 1 diabetes (T1D) diseased pancreas, and then apply this platform to address the gap in available methodology to take full advantage of T1D organ repositories such as nPOD in the effort to derive molecular profiles of individual -cells and cells of other types associated within the islet that are involved in the onset of silent T1D and disease progression. We will measure gene expression profiles from individual cells, and from this data identify biomarkers and gene expression phenotypes of silent T1D compared to more advanced stages of disease. We will exploit “big data” to apply pathway analysis to the gene expression results in order to identify putative molecular mechanisms of silent T1D and disease progression, providing a basis for the development of novel therapeutic approaches. We will test matched serum to identify useful biomarkers of silent T1D from this accessible tissue, and potentially to provide the capability to divide patients into subgroups based on prognostic outcome of whether disease will progress or not.
-
Bruce Seligmann
BioSypder Technologies, Inc
In Site TO-Seq Profiling of Gene Expression and Proteins in T2DWe will select T2D patients, among them subjects with DNA variants, and profile gene expression, expressed mutants, and proteins from sub-areas down to the level of single islets and single cells within archived pancreas FFPE tissues using a novel in situ TO-Seq assay coupled with immunohistochemistry staining of protein biomarkers, retaining the morphologic context of the profiling data. Among the set of genes measured will be biomarkers of every known molecular pathway in addition to genes associated with the development, presentation, dysregulation, and pathogenesis of T2D. By obtaining in situ gene expression profiles defining for all the cell types involved in diseased vs normal tissue within both their morphologic context and the context of genetic variants we will be able to identify molecular phenotypes and mechanisms and correlate these to cell-cell interaction, tissue homeostasis, and genetic variants. Because TO-Seq is a targeted sequencing-based assay that can measure thousands of genes, and multiplex many samples within a single sequencing run it will enable profiling data to be obtained from many separate cells at an affordable cost/cell in order to achieve the statistical power required to produce quality data supporting reliable interpretation and bioinformatics analysis. No such data exists today, and there is no system capable of producing such data on a large scale at an affordable price. Laser capture microdissection (LCM) can produce single cell profiles within their morphological context, but at a high cost which severely limits the number of samples that can be profiled. We provide preliminary data demonstrating excellent correlation and sensitivity of TO-Seq measurement of gene expression from matched fixed and frozen tissue and successful implementation of the in situ TO-Seq protocol and its integration with immunohistochemical (IHC) staining of antibody biomarkers. We demonstrate the feasibility of performing single (prostate) gland profiling with this assay. Based on these preliminary data we will optimize the in situ TO-Seq/IHC assay, implement a single islet and cell sampling system benchmarked against LCM, implement the content for the assay, and then profile the cell types present in pancreas of normal vs T2D, and between areas of pancreas exhibiting a normal vs T2D histopathology. From these data we expect to gain an understanding of the impact of genetic variant(s) at the level of gene (and protein) expression, cell interactions, and upstream regulation, and molecular mechanisms and phenotypes as they potentially contribute to disease and its progression.
-

David Serreze & Kevin Mills
The Jackson Laboratory
Targeting AID expressing B-lymphocytes as possible clinically applicable T1D interventionDiabetogenic B-lymphocyte activity may be enhanced by SHM induced increases in Ig antigen
affinity.
-SHM requires AID induced double strand DNA breaks repaired by RAD51 complex proteins.
-AID is expressed at higher levels in B-lymphocytes from NOD mice than control strains
-A small RAD51 blocking molecule specifically induces apoptosis of AID expressing B-lymphocytes
from both NOD mice and humans.
-RAD51 blockade is being tested as a new B-lymphocyte directed late disease stage T1D intervention
approach in NOD mice.
-For possible future clinical translation purposes it will be important to assess if diabetogenic Blymphocytes
in humans are undergoingAID induced SHM, and thus may be susceptible to depletion
by RAD51 blockade.
-To address this need nPOD is being requested to supply five cryopreserved PLN samples each from
T1D patients and controls.
-We will use these nPOD samples to determine if B-lymphocytes within PLNs of T1D patients express
higher AID levels than those from controls and thus more susceptible to RAD51 blockade induced
apoptotic death.
-Previous published work by Jax investigators demonstrate they have an ability to assess AID
expression by human B-lymphocytes, and determine their sensitivity to RAD51 blockade induced
apoptosis. -

Åke Sjöholm
Södertälje Hospital, Södertälje, Sweden
Ljungan virus in T1DIt was earlier reported the finding that the incidence of type 1 diabetes correlates with small rodent abundance in Sweden. This and other findings initiated an effort to isolate novel infectious agents from small rodents in an attempt to find an etiological agent for type 1 diabetes in humans. As a result of these efforts, a new picornavirus (Ljungan virus [LV]) was isolated from bank voles trapped in Sweden. Moreover, it has been shown that more than one-third of wild-caught bank voles and their offspring develop diabetes when kept under laboratory conditions. This was accompanied by changes in pancreatic islet structure, islet cells becoming immunohistochemically positive for LV and elevated levels of islet autoantibodies. The finding of LV in the pancreatic islets of the diabetic bank voles raised the general question whether or not LV can participate in the development of diabetes in rodents as well as in humans.
In pilot experiments using human pancreatic specimens from nPOD, we have found faint immunostaining for LV in two autoantibody-positive patients using a rabbit anti Ljungan virus protein 1 (LV VP1) antibody. Other type 1 diabetic patients, as well as non-diabetic controls, stained negative for LV in the pancreas. Due to the weak intensity of the immunostaining, results are inconclusive and not further pursued at this time.
-

Lydia Sorokin & Eva Korpos
University of Muenster, Germany
Peri-capsular basement membrane degradation during leukocyte penetration Into the pancreatic islet during development of human type 1 diabetes and tertiary lymphoid organs in human T1DSeveral steps are crucial for induction of type 1 diabetes (T1D); the first is extravasation of CD4+ T lymphocytes from blood vessels into the pancreatic tissue, the second is penetration of the peri-islet basement membrane (BM) surrounding the β-islets, and third the β-cell destruction which leads to appearance of disease symptoms. BMs act to separate tissue compartments and represent barriers to the movement of both soluble molecules and cells. Hence, cells penetrating such protein barriers must employ specialized mechanisms (1,2). The main question addressed in our nPOD project is what is the mechanism used by leukocytes to penetrate the peri-islet BM and, thereby, reach the insulin producing β-cells during the development of T1D, in the non-obese diabetic (NOD) mouse model and in recently diagnosed T1D patients.
We recently published a comprehensive analysis of the extracellular matrix (ECM) composition of peri-islet capsules, composed of the peri-islet BM and subjacent interstitial matrix, in development of T1D in NOD mice and in human T1D (3), for which nPOD samples were crucial. Our data demonstrated global loss of peri-islet BM components only at sites of leukocyte infiltration into the islet in both mouse and man. Stereological analyses reveal a correlation between incidence of insulitis and the number of islets showing loss of peri-islet BM versus islets with intact BMs, suggesting that leukocyte penetration of the peri-islet BM is a critical step in disease development. We identified cathepsin S, W, and C activity at sites of leukocyte penetration of the peri-islet BM in association with a macrophage subpopulation in NOD mice and human T1D (3). Interestingly, the peri-islet BM is reconstituted once inflammation subsides, indicating that the peri-islet BM-producing cells are not lost due to the inflammation, which has important ramifications to islet transplantation studies (3). We are now investigating how the cathepsins are involved in loss of the peri-islet BM: Studies are underway to detect and image cathepsin activity in mouse NOD and nPOD samples using cathepsin activity-based probes, which could lead to the development of novel diagnostic tools and therapeutic targets. In addition, we are examining the BM of the pancreas from different stages of human T1D and islet transplantation, with the aim of identifying ECM molecules that could promote islet survival.
Reference:
1) Korpos, E. et al, 2010. Cell Tissue Res. 339:47-57.
2) Wu, C. et al, 2009. Nat Med. 15:519-27.
3) Korpos et al, 2013. Diabetes 62:531-42.
Investigation of tertiary lymphoid organ formation in nPOD samples with ‘’typical insulitis’’
Lymphoid neogenesis is associated with chronic inflammatory diseases such as type 1 diabetes (T1D) and leads to formation of tertiary lymphoid organs (TLO). TLOs, like secondary lymphoid organs (lymph nodes and the spleen), are compartmentalized into T- and B-cell zones and contain reticular fiber networks composed of reticular fibroblast cells (RFCs) and reticular fibers (RFs), the latter of which are unique extracellular matrix (ECM) structures that have been characterized in our lab (Sixt et al, 2005; Song et al, 2013). The RFs of lymph nodes constitute the structural backbone of the organ, but also support the migration of T- and B-cells (Bajénoff et al, 2008) and conduct fluid transport of small molecules and soluble antigens (<70kDa), (Sixt et, 2005). Although TLOs have been described in many chronically inflamed tissues, their characterization from the ECM point of view is limited and their contribution to autoimmune diseases such as diabetes is not clear.
Using whole mount stainings and confocal microscopy we have characterized the cellular and ECM composition of RFs in TLOs of the inflamed pancreas of non-obese diabetic (NOD) mice, where the autoimmune destruction of insulin producing β-cells leads to replacement of pancreatic islets by TLOs. Our data suggest a role for the RFs in the conduction of chemokines from the infiltrating front to the high endothelial venules (HEVs) to propagate immune cell recruitment to the inflamed islet as well as antigen presentation to dendritic cells bound to the RFs, thereby, accelerating and promoting the inflammatory response. To be able to stem this acceleration step by interfering with TLO formation represents a potential mode of at least slowing T1D progression. However, it is first necessary to define whether the structure of the TLO is the same in mouse and in human T1D pancreatic samples with “typical” insulitis.
To investigate whether the presence of TLO in nPOD samples is correlated with insulitis we will perform immunofluorescence staining using antibodies specific for 1) different ECM molecules to identify the structure of the RFs; 2) HEVs, immune cell compartments and reticular fibroblasts to define the interconnection between the infiltrating front and the HEVs; and 3) auto-antigen (insulin) and chemokines to define whether the RFs also have a conduit function.
Reference
1) Sixt M, et al, 2005. Immunity, 22(1):19-29.
2) Bajénoff M, et al, 2008. J Immunol, 181(6):3947–3954.
3) Song J, et al, 2013. Proc Natl Acad Sci, 110(31):E2915-24.
-

Stephan Speier, Alberto Pugliese
Helmholtz Zentrum München, Germany
Funded by: Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
Development of a platform for ex-vivo functional and 3D morphological assessment of islet physiology in nPOD human pancreata, also in response to viral infections associated with type 1 diabetes pathogenesisImpairment and destruction of beta cells by an autoimmune attack leads to the onset of hyperglycemia in type 1 diabetes (T1D). However, knowledge on changes in islet cell physiology leading to dysfunction, failure and death of the cells is incomplete. This is partially due to technical limitations which do not allow the study of islet function within intact pancreatic tissue, but rely on the enzymatic isolation of islets. During isolation islets suffer from enzymatic and mechanical stress. After isolation the islets are separated from their surrounding tissue, including most of the infiltrating cells. In addition, enzymatic isolation of structurally damaged islets is challenging and hampers the study of islet physiology in T1D.
Our project aims to establish a pancreas tissue slice platform for the study of human islet physiology in the pathogenesis of T1D. Pancreas tissue slices allow the study of islet physiology in an intact environment in situ, avoiding isolation stress and separation from their surroundings. In rodents pancreas tissue slices have been successfully used to study rodent islet biology under various conditions. We recently established the pancreas tissue slice platform using human pancreas. We plan to establish this technique at the Diabetes Research Institute, University of Miami, and at the nPOD Organ Processing and Pathology Core, University of Florida. This will enable the use of pancreas tissue slices for the ex-vivo study and functional assessment of nPOD pancreata. Furthermore, we aim to develop an organotypic culture platform to allow investigating human islets within intact pancreatic tissue over several days. Finally, we will assess the feasibility of this culture platform by using exposure to viruses as a model system to explore effects on islet cell pathophysiology. We believe combination of this unique network with our novel approach will facilitate new insight into the pathogenesis of diabetes. Of note, this platform will empower additional studies to be conducted, beyond those described here. This effort is conceived as a collaborative working group effort which will seek support from the Helmsleüy Charitable Trust George Eisenbarth nPOD Award for Team Science.
-

Jo Spencer
King's College London
Funded by: Breakthrough T1D
Phenotype, specificity, diversity and clonal spread of B cells in T1DMIn this preliminary screening of tissues, we propose to characterize B cells in the pancreas of cadavers that are autoantibody positive. Pancreas of non-autoantibody positive cadavers will be studied as controls. We propose to use immunohistochemistry to analyse cell surface marker expression, together with an analysis of the microantomical distribution of B cells and their relationship to other cells. We will attempt to locate specific antibody producing cells. We will be particularly interested in a population of large B cells with dendritic processes that often connect with T cells. These cells, which are normally located in thymus and in T cell zones of organized lymphoid tissues may be antigen presenting B cells.
-

Roland Stein
Vanderbilt University Medical Center
Funded by: NIDDK
Analysis of Islet Enriched Transcription Factor Expression and Activity in Normal & Type 2 Diabetic IsletsThe therapeutic treatment of type 1 and type 2 diabetes mellitus (T1DM, T2DM) has vastly improved over the past decades; however complete normalization of glucose remains elusive without proper functioning pancreatic β cells. Truly, sustained and effective treatment of T1DM and T2DM must involve retaining/replacing the function of pancreatic β cells. To this end, we must understand the molecular mechanisms that govern β cell development and function along with understanding the molecular progression of the disease.
The Stein lab and others have focused on the large Maf transcription factors, MafA and MafB, and their role in the β cell. Loss of MafB impedes normal β cell development in mouse models whereas loss of MafA restricts normal adult β cell function. In recent published data, we have shown that MafA and MafB are lowered in db/db mouse models and T2DM donor samples. Collectively, this data reveals the importance of MafA and MafB in normal β cell function and presents an area of research that needs to be further examined.
Using nPOD, we are currently characterizing transcription factor levels and investigating coregulator interactions in T1DM and T2DM donor samples. This data is crucial to the understanding of the human β cell situation and vital for β cell replacement therapy protocols.
-

Terry Strom, Thomas Thornley & Maria Koulmanda
Harvard Medical School
To identify mRNA and miRNA biomarkers associated with exosomes that predict the onset of T1DThere are currently no clinically precise biomarkers that predict the development of type 1 diabetes (T1D). This limits the ability to identify patients who will become diabetic at a time when they have a greater beta cell mass and thus may respond more favorably to interventional therapeutics aimed at stopping T1D progression. To this end, we propose to identify biomarkers that predict T1D progression by examining exosomes in at-risk and nondiabetic control individuals. Exosomes are small particles released by cells during inflammation that communicate “danger” to other cells nearby and at a distance. Exosomes contain numerous mRNA and miRNA messengers that alter the biology of target cells, which include cells of the innate and adaptive immune systems. We propose to characterize the transcriptional (mRNA and miRNA) contents of exosomes from at-risk and non-diabetic control tissues in order to identify potential biomarkers that predict progression of T1D.
-
Jonathan Sweedler, Mark Atkinson, and Clive Wasserfall
University of Illinois
Using Emerging Techniques to Identify Novel Islet Signatures in Type 1 and Type 2 DiabetesThough the pancreatic islets are known to play critical roles in both Type 1 diabetes (T1D) and Type 2 diabetes (T2D), the distinct mechanisms are poorly understood. Recent findings demonstrate that chemical heterogeneity exists within the classic islet endocrine cell types, even in the normal/healthy pancreas setting. For example, there are subtypes of beta cells with distinct transcriptomes that respond differently to chemical signals. One critical scientific question is how does this cellular heterogeneity influence chemical signaling in T1D and T2D? We hypothesize that characteristic changes in chemical heterogeneity within the various human islet cell types during diabetes progression alter the signaling pathways inside and outside of the pancreatic islets to potentiate disease. Determining the dynamics of these changes will provide insight into chemical mechanisms that can be exploited for the prevention and treatment of T1D and T2D. Hence, there is a need for high-throughput and highly sensitive technologies capable of probing a sufficient number of chemical constituents in single cells. We propose three unique measurement platforms that implement advanced single cell mass spectrometry and analyte separation techniques to characterize the peptide and small-molecule content in individual human islet cells affected by T1D and T2D: 1) high-throughput single cell matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS); 2) quantitative capillary electrophoresis (CE)-MS with a focus on small molecule metabolites; and 3) CE-laser induced fluorescence (LIF) and liquid chromatography (LC)-MS characterization of the levels of D-amino acids. We expect that this research will dramatically improve the understanding of the cellular mechanisms of both T1D and T2D pathogenesis, and elucidate new chemical parameters characteristic of each disease, helping to identify novel pathways for therapeutic intervention.
-

Gerald Taborsky, Jr.
University of Washington
Funded by: NIH
Glucagon secretion and islet neuropathyTo confirm and publish our preliminary observations (Diabetes 55 (supplement 1):64-OR, 2006) suggesting that humans with Type 1 Diabetes Mellitus have a marked and islet-selected loss of their sympathetic nerves, similar to that we have observed in animal models of autoimmune diabetes. If true such a defect may contribute to the impaired glucagon response to induced hypoglycemia seen early in Type 1 diabetes.
-

David Taylor-Fishwick
Eastern Virginia Medical School
The patterns of NADPH oxidase-1 expression in the course of human diabetesIncreased levels of reactive oxygen species (ROS) lead to dysfunction and subsequent loss of insulin-producing beta cells in pancreatic islets. Several lines of evidence support the importance of increased NADPH oxidase (NOX) activity as one of the contributing factors to elevated levels of ROS in beta cells. NOX-1 activity has been established as one of the sources of ROS in beta cells under experimental diabetic conditions. A contribution of NOX-1 to beta cell dysfunction in human diabetes is supported by preliminary studies. To further validate these observations as a first step we seek to establish the pattern of expression NOX-1 in normal and diabetic human islets. We hypothesize that NOX-1 will be elevated in diabetic islets. NOX-1 expression will be studied in relation to both the specific cells within pancreatic islets (beta cells, alpha cells) identified using well established specific protein markers for each phenotype as well as to the endocrine “sufficiency” status of such cells. In order to specifically examine qualitatively and quantitatively islets from 6 non-diabetic controls, 6 type 2 diabetes, 6 type 1 diabetes of short and 6 of long duration as well as 6 autoantibody positive would be employed; hence a total of 30 donors will be included in this study. Paraffin embedded 7 ìm thick sections of pancreas will be immunostained, using a protocol previously developed specifically for nPOD specimens. Commercially available antibodies raised against human NOX-1, insulin and glucagon will be used. Stained sections will be subjected to microscopic evaluation using both conventional fluorescent microscope and confocal microscope. A qualitative and quantitative analysis of the obtained images will be employed. We expect that an increase in expression NOX-1 will be observed in islets obtained diabetic and autoantibody positive donors.
-

Charles Thivolet
University Claude Bernard Lyon 1
Characterization of new cell markers associated with beta cell failure in diabetesThe percentage of reduction of beta cell mass in patients with Type 1 diabetes (T1D) of recent onset cannot fully explain the reduction of endogenous insulin production that leads to hyperglycemia. In addition, functional defects of the beta cells may prevail from reduction of beta cell mass during Type 2 diabetes. In boty situation chronic inflammation may explain why beta cells cannot produce sufficient amounts of insulin to cope with insulin demands. The purpose of the project is to examine in situ both the mitochondria and RE-mitochondria interactions, key organelles of beta cells. Apart from being involved in the capacities of beta cells to secrete insulin upon demand, mitochondria have a central role in beta cell fate through ROS generation, capacities to transfer apoptotic signals and in the orchestration of inflammatory responses and inflammasome activation. The levels of ER-mitochondria overlap and number of ER-mitochondrial associated membranes (MAM) will be quantified using an in situ proximity ligation assay. We plan to compare the degree of RE-mitochondria interactions in inflamed vs intact islets, in beta vs non beta cells, in patients with T1D vs T2D. Expression of specific mitochondrial proteins involved in Ca2+ uptake and in the balance of mitochondria fission and fusion processes will be analyzed. Since excessive production of ROS and release of oxidized mitochondrial DNA following mitochondrial stress can be sensed by the inflammasome, levels of NLRP3 expression will be assessed both in mononuclear cells contained in inflamed islets and eventually in the beta cells. This could be an important link between innate immunity in response to viral infections and autoimmunity to beta cells. Since miRNAs can also regulate mitochondria activity, redox state and inflammatory pathways, semi-quantitative analysis of specific miRNAs will be performed by fluorescent micro-RNA in situ hybridization (FISH) technique with digoxigenin-labeled probes. Altogether, this project will give a comprehensive analysis on the role of mitochondria in the beta cell defects during the early phases of diabetes.
-

Ranjeny Thomas & Katharine Irvine
University of Queensland
Funded by: Breakthrough T1D
Pathological changes associated with chronic RelB activation in T1DDendritic cells (DCs) play a crucial role in establishing and maintaining the balance between immunity to pathogens and tolerance to self. Whether DCs evoke T cell activation or tolerance in response to antigen presentation is determined by the physiological context in which they differentiate and mature. DC differentiation and inflammatory signalling is abnormal in type 1 diabetes (T1D), but the relationship between this abnormality and disease pathogenesis requires clarification. We have shown constitutive activation and defective inflammatory responses in critical sub-units of the NF-kappaB transcription factor family, known as RelB and p65, which are required for myeloid DC differentiation, in T1D monocytes and DCs. Abnormal RelB activation may constitute a novel T1D risk phenotype. Our data indicate that peripheral blood cell RelB activation and associated inflammatory gene expression is increased in autoantibody-negative first-degree relatives of T1D patients. In autoantibody-positive and autoantibody-negative subjects, serum inflammatory cytokine expression is heterogeneous. We hypothesize that premature RelB activation and pro-inflammatory cytokine production during DC differentiation compromises the development of immunological tolerance and contributes to T1D pathogenesis. In this project we wish to investigate whether RelB activation and inflammatory cytokine expression in peripheral blood is associated with distinct pancreatic pathology in T1D and at-risk subjects. This information could enable us to stratify pancreatic pathology in living T1D patients and at-risk subjects, based on their peripheral blood RelB and inflammatory biomarker status. Moreover strategies for early therapeutic intervention to prevent or treat T1D may differ depending on pancreatic pathology.
-
John Todd, Vincent Plagnol & Herbert Virgin, IV
University of Cambridge, UK
Funded by: Breakthrough T1D
Viral sequence discovery by high throughput DNA sequencing of the nPOD diabetic pancreatic samplesSeveral lines of evidence, in particular genetic association data of antiviral response genes with T1D risk, point to a role of viral infections in T1D etiology. However, multiple questions remain about the causal mechanisms and the type of viruses implicated. To contribute to these research questions, we are using high throughput sequencing technologies to find traces of viral RNA in the nPOD pancreatic organ donor T1D cases. Our strategy is based on deep RNA sequencing combined with sequence enrichment for known viral sequences to maximize the sensitivity of viral sequence detection. We then use bioinformatics tools to separate human and viral reads, characterise these reads and eventually provide a description of the virome of nPOD samples.
-

Antonio Toniolo, Andreina Baj & Roberto Accolla
University of Insubria Medical School, Italy
Funded by: CARIPLO Foundation
Detection of enteroviruses in lymphoid tissue of donors with T1D of short duration and attempts to identify the infected cell type(s)If type 1 diabetes (T1D) is caused/triggered by a viral infection in select genetic backgrounds – and Enteroviruses (EVs) are felt as major culprits – investigators are expecting to find EVs into pancreatic Langerhans islets and also in association with spleen, lymph nodes, peripheral blood leukocytes. Infection of lymphoid cells, in fact, is common in systemic infections. Using an original method that couples virus amplification in susceptible cells with RNA amplification assays capable of detecting persistent and mutated EV types, we showed that different EV species are present in blood leukocytes of two thirds of children/adolescents at the clinical onset of T1D. Initial study of living cells from nPOD cases (T1D, non-diabetics) demonstrated that EV genomes and infectivity are present also in spleen and lymph nodes of T1D cases (1 to 30 years post-diagnosis). Thus, EVs appear to produce chronic silent infections in diabetes as in the post-polio syndrome (that is being studied by our group). If the viral genome is present, then virus proteins should also be detectable. Initial work done with colleagues looking at the same nPOD samples by ISH, immunohistochemistry, proteomics showed a strong concordance between genome detection and protein expression. So far, we have been successful in sequencing short stretches of RNA encoding conserved EV enzymes. We failed, however, in detecting genes encoding EV capsid proteins. This may be due to the pronounced variability of these proteins, especially in chronic infections. An additional aim of our studies is to determine which leukocyte subsets are carrying EVs. Preliminary results suggest that T cell subsets may be infected. Evidence in this direction, would push the idea that virus-induced immune disturbances might contribute to autoimmunity in T1D. Proof that chronic EV infections are indeed associated with T1D has not been reached, however collaborative studies of the same nPOD cases using many different approaches will certainly reveal if the virus hypothesis is a viable one. Should EVs be implied, major implications for human health will ensue (antivirals, novel preventive measures, etc.).
-

Steven Tracy
University of Nebraska
Enteroviral infection and T1DDespite convincing evidence linking human enteroviruses (HEV) with type 1 diabetes (T1D) onset, it is not known whether certain HEV are the ones which are most commonly associated with T1D onset or if T1D onset is associated with any HEV serotype.
-

Emil Unanue
Washington University School of Medicine
Funded by: Breakthrough T1D
The immunogenicity of abnormal catabolites of insulin metabolismWe are searching for abnormal products of insulin metabolism that appear in beta cell as a result of the increased demands for insulin. These abnormal catabolites in NOD mice select for very unique CD4 T cells that initiate this chronic autoimmune reaction. The plans are to search for these abnormal products in islets from early diabetic subjects. Their presence in human islets may point to key antigens that may be the target of future immunotherapy.
-

Hans-Dieter Volk, Alexandra Mourad & Matthias von Herrath
Charité - Universitätsmedizin Berlin
Funded by: BMBF, Graduate School DFG, Einstein Visiting Fellow
Mass Spectrometry Based Digital Histology nPOD (Berlin BCRT)The individual heterogeneities of Type 1 diabetic pancreas could hinder a successful investigation and classification of the underlying mechanisms in diabetic disorders. This indicates the necessity of in situ analysis of tissue sections by novel imaging mass spectrometry technology, which enables the spatial investigation of protein profiles from an examined tissue section, thus allowing to relate and to visualize changes in tissue histology to changes in the proteomic signature of that tissue. Through this systemic technique we will be able to understand the pathological cause of the changes during early stages of Type 1 diabetes, which could give us more insight into pinpointing preventive strategies for this disease.
-

Matthias von Herrath and Teresa Rodriguez
University of California, San Diego
Funded by: NIH
In situ detection of pro-inflammatory cytokines within pancreatic islets from diabetic subjectsIn Type 1 Diabetes (T1D), the complex interplay of immune cells shifts the balance in favor of the development of lymphocytes, that attack structures of our own body: autoimmunity. These attacks resulting in an inflammation in the pancreas and, eventually, to the destruction of insulin producing beta cells.
Cytokines are small proteins that are released by cells and affect the behavior of other cells. They are oftentimes involved in promoting inflammation. We believe, that a network of “cytokines” determines the direction of the immune response towards the destruction of beta cells, and, as recent findings suggest, that this inflammatory milieu precedes the onset of the disease. However, it remains elusive so far, which cytokines are produced and when, and how they interact in such a complex network to sustain inflammation.
In this project, we use highly sophisticated chemical and bioinformational tools to detect important cytokines that are known to promote inflammation: Interleukin 1beta, Interleukin 6, and Tumor Necrosis Factor alpha in tissue pieces from diabetic patients, who have donated their pancreas for research.
This allows us to analyze both the presence of cytokines as well as their interactions during the very early days of diabetes at the single cell level and to create a so called “cytokine map” of the disease. Knowing which cytokine is released by which cell and when is a first step towards the development of very specific therapies for the treatment of diabetes and of other autoimmune diseases.
-

Thomas Waldmann & Jing Chen
NCI/NIH
Funded by: Metabolism Branch, NCI, NIH, DHHS
The role of IL-15 and IL-15Ra in the pathogenesis of type 1 diabetesInterleukin-15 (IL-15) is a pro-inflammatory cytokine that promotes the activation and maintenance of natural killer (NK) and CD8 (+) T-effector memory (T-EM) cells. In patients with type 1 diabetes, elevated serum levels of IL-15 have been reported. Using an assay developed in the lab, we demonstrated that the serum sIL-15Rα levels were significantly higher in patients with T1D compared with those of normal controls. To investigate whether islet overexpression of IL-15 and IL-15Rα could play a role in the pathogenesis of T1D, we generated double transgenic mice with β cell specific expression of both IL-15 and IL-15Rα under a rat insulin promoter. The mice developed hyperglycemia, marked mononuclear cell infiltration, β cell destruction and anti-insulin autoantibodies that mimic the early events of human T1D. Inhibiting IL-15 signaling with anti-IL2/IL15Rβ (anti-CD122), which blocks IL-15 transpresentation, or with the Janus kinase 2/3 inhibitor tofacitinib, reversed the diabetes in the transgenic mice. This suggested a causative role of IL-15 and IL-15Ra in the induction of insulin dependent diabetes in mice.
To further investigate whether IL-15 and IL-15Ra play a role in the pathogenesis of human type 1 diabetes, we investigated the expression of IL-15 and IL-15Ra in the islets of patients with type 1 diabetes, anti-insulin autoantibodies and normal donors. Immunohistochemistry demonstrated IL-15 expression in the islets in all T1D patients examined whereas IL-15 was detected only in the pancreatic islets in one out of four normal donors and two out of five autoantibody positive donors. Interestingly, in two cases where IL-15 was positive, we also detected islet expression of MxA, a GTPase induced by type 1 IFN (α/β) that has been reported to interfere with virus multiplication and spread. Furthermore, IL-15 and IL-15Rα mRNA levels were also higher in the islets from T1D patients compared to that from autoantibody positive donors or normal donors. The islets were microdissected based on insulin positivity using laser capture microscopy (LCM). Taken together, our data suggest that disordered IL-15 and IL-15Rα may be involved in the pathogenesis of human T1D.
-

Shannon Wallet
East Carolina University
Funded by: NIH/NIDDK
Role of mucosal epithelium in autoreactivityOne of the major questions we still don’t have an answer to is why some people who are genetically predisposed for development of T1D don’t progress and why others do. In addition, why there are different timings for the progression of this disease (i.e. why are some faster than others). Our thoughts are that these differences are due to differences in environmental experiences. It is important to note that we feel that these different environmental experiences can be big or small (i.e. living in different areas, eating different foods, or infection with different organisms). Thus, my laboratory studies the communication of the immune system with the environment at the largest environmental interface within our body, the gastrointestinal track. We feel that if we can understand the difference in this communication that occurs in T1D and how it shapes the immune system, we can not only figure out the initiating events of this disease process, but design better therapies and even preventive treatments to curb the disease process.
-
Shannon Wallet
East Carolina University
Characterization of Mesencymal Stem Cells in T1DThe development of MCS-based therapies for autoimmune diseases, including T1D, is a new field with a lot of promise, but also a lot of uncertainty. While successful treatment of disease allogeneic MSCs has been achieved, some MSC treatment failure has also been reported. The significance of this proposal is that it will fill a significant gap in the current understanding of mechanisms associated with human MSC-based therapies for T1D. These studies raise critical issues such as which patients could benefit from MSC treatment and when autologous or heterologous MSCs are best to be utilized. Here we propose to investigate whether T1D-derived MSCs are as effective in suppressing inflammation as non-T1D derived MSCs. Importantly, we are proposing to evaluate this using human samples as there is murine data to suggest that T1D-derived MSCs are genetically altered which affect their efficacy as immunosuppressive therapies.
-

Mark Wallet & Robert Hromas
University of Florida
Funded by: ADA
Role of CCL21 in Directing Immune Invasion of Pancreatic IsletsType 1 diabetes results, at least in part, from invasion of pancreatic islets by T lymphocytes and antigen presenting cells (APCs) including macrophages and dendritic cells (DCs). What drives the migration of these immune cells into the islets remains unknown. A key step in the arrival of immune cells to a specific location is adhesion to the inner lining of blood vessels and then transit through the vessel wall into the surrounding tissue. Chemokines are important proteins in this process. Chemokines that are expressed by the cells making up blood vessels alert immune cells to nearby inflammation and assist the immune cells with leaving the vessel. One chemokine with this function is CCL21. Activated APCs and naïve T cells express the surface receptor CCR7 that binds CCL21 and directs the cells towards environments where CCL21 is present. We found through profiling of genes expressed in the islets of T1D subjects in the nPOD cohort that CCL21 levels are elevated in the islets that have been infiltrated by T cells but not islets lacking T cells. We hypothesize that endothelial cells of the islet are expressing CCL21 and that drugs that block CCL21 activity can prevent destructive immune invasion of the islets. This study is designed to determine where in the islet environment CCL21 protein is found. Since immune infiltrates are focal, we anticipate that CCL21 will co-localize with regions of the islet where APCs or naïve T cells have infiltrated.
-
Clive Wasserfall
University of Florida
Humoral immunity in type 1 diabetesOur project entitled humoral immunity investigates several serological aspects of Type 1 Diabetes (T1D). Our group also serves as the ELISA autoantibody core wherein we oversee the screening laboratories and together with the University of Colorado maintain quality assurance and control for this program. However we also conduct research into additional aspects of autoantibodies from nPOD such as Islet cell autoantibody assays on autologous tissue. From this the idea of looking for complement deposition in pancreas tissue of autoantibody positive donors as well as those with T1D arose. We published the findings in Diabetes Care and in sum we found that C4d deposition was mainly associated with blood vessels and the extracellular matrix that surrounds blood vessels an the pancreatic ducts. This was most pronounced in T1D pancreas tissues and was interpreted as evidence of ongoing inflammation of the pancreas in this disease.
-

Hongju Wu & Vivian Fonseca
Tulane University
Pancreatic GLP-1 vs glucagon production in healthy and diabetic patientsGlucagon-like Peptide 1 (GLP-1) and glucagon shares the same precursor molecule proglucagon, but each arises from a distinct posttranslational process in a tissue-specific manner. Recently GLP-1 has been shown to be co-expressed with glucagon in pancreatic islet cells. Our preliminary data showed GLP-1 was progressively up-regulated in pancreatic islets during type 2 diabetes development. These data suggest intra-islet GLP-1 production may have an impact on the clinical and pathophysiological processes of these diseases and may be a target for therapeutic approaches. This project is thus designed to investigate this matter in human type 1 and type 2 diabetic patients. In addition, since GLP-1 based drugs, incretins, have been developed and approved for clinical use, it is thus essential to investigate their impact on intra-islet GLP-1 production. The main objectives of this project are thus 1) To investigate whether GLP-1 vs glucagon production in pancreas of human has changed with the development of type 1 and type 2 diabetes; and 2) To investigate whether the GLP-1 based therapies (i.e., incretin therapy) had any impact on the relative production of GLP-1 vs glucagon. To accomplish the goals, pancreatic tissues from normal, type 1 diabetic, type 2 diabetic without incretin treatment and type 2 diabetic with incretin treatment will be analyzed by immunofluorescence staining. The GLP-1 vs glucagon expression will be quantified and analyzed for statistical significance. In addition, the matching blood samples will be used to obtain physiological data related to glucose regulation. The results will help us understand the complete effect of incretin drugs, and identify potential safety issues. Therefore, the study will not only advance our knowledge on the regulation of intra-islet GLP-1 production in diabetes, but also have significant impact on the clinical practice regarding current use of incretins.
-

Qibin Zhang & Mark Atkinson
The University of North Carolina Greensboro
Human pancreatic tissue proteome of type 1 diabetesThe goal of this project is to comprehensively profile the proteins expressed in human pancreas within both the islets and their surrounding environments, and to obtain relative expression levels of pancreatic proteins from different organ donor groups using mass spectrometry based bottom-up and top-down proteomics technologies. Snap-frozen pancreatic tissue sections from organ donor groups with 1) autoantibody negative, no diabetes; 2) autoantibody positive, no diabetes; 3) autoantibody positive and T1D; and 4) autoantibody negative and T2D will be analyzed on both the peptide and intact protein levels for comprehensive identification and quantification of proteins and protein isoforms that differentially expressed among these subject groups. To complement with our study on pancreatic tissue sections, islets and beta cells will be isolated from pancreatic tissues using laser capture microdissection for their comprehensive proteomic characterization and comparison as well. The significantly changed proteins and the specific pathways and networks they are involved in may serve as candidate biomarkers for diagnosis and aid in elucidation of the pathogenesis of T1D.
-

Emmanuel Zorn, Yufeng Shen, Ivan Gerling
Columbia University
Funded by: Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science
Immunoregulation of type 1 diabetesType 1 Diabetes (T1D) is a disease that results from the autoimmune destruction of insulin-producing beta cells in the pancreas. The current treatment for T1D does not cure the disease or prevent the possibility of other serious complications. The mechanism that leads to the loss of immune tolerance still remains unclear; scientists believe that both genetic and environmental triggers play a role, making it challenging to develop treatments that address both. Investigators at the Columbia University Center for Translational Immunology (CCTI) conduct many research studies aimed to determine factors involved in T1D. The purpose of our study is to investigate genetic variants limited to immune dysregulation in T1D through next generation sequencing of genetic material from tissues of T1D donors. Elucidating the genetic factors involved in T1D will contribute to our knowledge of this disease and autoimmunity.